
基于二水草酸镁(MgC2O4.2H2O)的无水碳酸镁(MgCO3)的高压制备和表征∗
2017-07-31 05:59梁文1李泽明1王璐颖1陈琳1李和平1
物理学报 2017年3期
梁文1) 李泽明1)2) 王璐颖1)2) 陈琳1)2) 李和平1)
1)(中国科学院地球化学研究所,贵阳 550081)2)(中国科学院大学,北京 100049)(2016年9月22日收到;2016年10月11日收到修改稿)
专题:高压下物质的新结构与新性质研究进展
基于二水草酸镁(MgC2O4.2H2O)的无水碳酸镁(MgCO3)的高压制备和表征∗
梁文1) 李泽明1)2) 王璐颖1)2) 陈琳1)2) 李和平1)†
1)(中国科学院地球化学研究所,贵阳 550081)2)(中国科学院大学,北京 100049)(2016年9月22日收到;2016年10月11日收到修改稿)
无水碳酸镁因为其重要的研究价值和广泛的应用而备受关注,因此探索无水碳酸镁的制备方法已经成为材料加工的一个重要课题.以二水草酸镁为起始原料,使用高温高压反应法成功地合成了高纯度的无水碳酸镁,对所得样品分别进行了粉末X射线衍射和Raman光谱的表征.通过热重分析以及尝试不同合成条件,给出了二水草酸镁-碳酸镁P-T相图并解释了高温高压合成碳酸镁的原理.利用偏光显微镜观察样品碳酸镁形貌、晶粒度以及晶体解理面,同时对样品的单晶粒做微区电子探针分析,给出了样品的镁含量.
无水碳酸镁,高压合成
1 引 言
无水碳酸镁(MgCO3)作为一种功能材料因其应用广泛而备受关注[1−6].在工业上,碳酸镁是一个重要的原料,主要用于制药、食品添加剂、工业橡胶等制造业.近些年来,碳酸镁作为一种新型的无机阻燃剂引起了众多研究者的重视[7,8].与传统的有机卤系阻燃剂相比,它具有无毒、无腐蚀性、对环境无污染等优点.同时,与无机无卤阻燃剂中的重要类型——氢氧化铝和氢氧化镁相比,无水碳酸镁不仅具有热分解速度快、吸热降温、吸热热容大等优点,而且还能够分解释放大量的二氧化碳隔离空气,起到有效的灭火作用.因此,无水碳酸镁是目前已报道的最好的阻燃剂材料.
在地球科学领域,碳酸镁作为一种矿物组成被称为菱镁矿,它对地球碳循环尤其是深部碳循环的研究有着极其重要的地球物理学和地球化学意义.在地球内部尤其是下地幔环境,碳的存在形式主要为碳酸盐、碳化铁、金刚石等,其中菱镁矿被认为是在下地幔的温度压力条件下最为稳定的碳的主要储备形式[9−13].研究表明,在下地幔存在的菱镁矿往往和菱铁矿(FeCO3)类质同象形成铁菱镁矿(FexMg1−xCO3),而铁菱镁矿在高温高压下的自旋相变对下地幔弹性、地震波波速的研究以及建立下地幔模型有着重要的价值[14−16].
然而,目前市场上的碳酸镁产品除了天然的菱镁矿以外,全部都是水合碳酸镁(MgCO3.3H2O)或者水合碱式碳酸镁(x MgCO3.y Mg(OH)2.z H2O),几乎没有现成的无水碳酸镁产品出售.而天然的菱镁矿总是含有不同程度的Fe2+和Mn2+等杂质,难以达到分析纯试剂的标准,无法满足实验研究所需样品的高质量、高纯度的要求.
尽管在常规条件下,无水碳酸镁的制备相当困难,但由于其重要的研究和应用价值,探索无水碳酸镁的制备方法已经成为材料加工的一个重要课题:在实验室尝试开发无水碳酸镁新的制备方法并改进其制备工艺,获得高纯度、高结晶度和高质量的碳酸镁样品,以便满足实验研究的各种需要.
在过去的研究中,已经报道了无水碳酸镁的一些不同的制备方法[17−19].尽管这些方法能够成功地获得纯度较高的碳酸镁样品,但还是存在一些明显的不足,比如,样品的结晶程度很差,晶粒尺寸太小无法满足微区测试和表征的要求,以及实验操作和条件控制复杂等.因此,在保证碳酸镁样品高纯的前提下,尽可能地提高样品的结晶程度并且改善合成工艺也是一个值得考虑的重要问题.
本文报道一种利用高温高压固相反应制备无水碳酸镁的新的方法.通过粉末X射线衍射(XRD)表征,所得到的样品为MgCO3单相,不含其他杂质.通过电子探针分析,得到了样品单晶粒微区的镁含量信息.利用偏光显微镜观察样品的形貌,样品的结晶程度较好,最大晶粒尺寸达到了200µm.更重要的是,在偏光显微镜下,特别清晰地看到了碳酸镁晶体的菱形解理面,这是三方晶系方解石型材料的自然解理(10¯11)面.这个重大的突破意味着,高温高压固相反应法将为进一步开展无水碳酸镁高质量的单晶的生长提供了必要的实验基础和技术保障.此外,该方法简化组装并使用低成本耗材,大幅降低了实验成本,同时控制反应时间在1 h以内,有效地提高了材料的合成效率.
2 实验过程
通过在高温高压下的固相反应获得碳酸镁,其过程如下:使用分析纯的二水草酸镁(MgC2O4.2H2O,99.9%)作为起始原料,使用压片机将草酸镁粉末压成圆柱形(Φ6 mm×3 mm),用银箔(0.025mm厚度)将其包裹做成样品.以NaCl为传压介质,石墨为加热器,内置K型镍铬-镍硅热偶(由于此高压实验组装使用了NaCl等低成本耗材,大幅降低了实验成本).将样品置于高压组装块中,在六面顶压机中进行高温高压反应,反应时间为1 h.为了讨论碳酸镁在P-T相图中的相稳定区域,我们尝试了0.5,1.0和1.5GPa压力下不同的温度条件.实验完成后,将获得的样品取出,清除样品表面银箔,即可得高压样品.
对获得的高压样品进行了粉末XRD表征,对XRD结果进行定量分析,结果见图1(a).对碳酸镁样品进行Raman散射表征,结果见图2.为了深入地解释碳酸镁在高压固相反应的合成原理,我们对原料二水草酸镁和合成的碳酸镁做了常压下的热重(TG)分析,结果见图3(a)和图3(b),同时,尝试了0.5,1.0和1.5 GPa压力下不同的反应温度条件,最终在P-T相图上给出了碳酸镁在高温高压下稳定的相区,结果见表1和图4(a).通过分析1.0 GPa压力下不同合成温度获得的碳酸镁样品的XRD主峰(104)的相对峰强((104)峰强/(113)峰强)和半高宽,给出了这组样品结晶度的最佳温度条件,结果见图4(b).图5(a)和图5(b)给出了制成的碳酸镁样品在偏光显微镜下的形貌照片.为了给出碳酸镁的微区成分,对碳酸镁薄片做了微区电子探针分析,结果见表2和图6.

图1 (a)高压合成产物碳酸镁的粉末XRD图谱;(b)MgCO3的三方晶系的晶体结构图Fig.1.(a)Powder X-ray d iff raction for the sample MgCO3synthesized at 1.0 GPa and 800◦C for 1 h,using Si(99.999%)as zero-off set calib ration;(b)MgCO3rhombohed ral structure.
3 结果与分析
3.1 粉末XRD结果
图1(a)为高温高压反应后获得的碳酸镁的粉末XRD图谱,样品合成条件为1.0GPa,800◦C,1h.衍射角2θ范围为10◦—80◦,所有的衍射峰都可以按照MgCO3的标准谱进行指标化,其晶体结构为三方晶系,空间群为R¯3c(no.167),结构如图1(b)所示.使用单晶硅粉末(99.999%)为XRD的零点矫正标准,对所有的衍射峰进行高斯-洛伦兹函数峰位置拟合,结构精修计算所得晶格参数为a=4.6365(8)Å,c=15.0262(11)Å,单胞体积为279.7Å3,理论密度为3.003 g/cm3.
3.2 Raman散射结果
图2为高温高压反应后获得的碳酸镁样品的单晶粒的Raman散射图谱,样品合成条件为1.0 GPa,800◦C,1 h.以单晶硅(99.999%)的520 cm−1峰为零点矫正,在波数范围200—2000 cm−1内,我们观察到6个Raman散射峰,分别位于210,326,737,1093,1444和1760 cm−1.对散射峰的位置用洛伦兹函数进行了拟合,根据已经报道的三方结构方解石碳酸盐一族(方解石族,包括CaCO3,MgCO3,FeCO3,MnCO3等)的Raman振动模式的研究[20,21],6个Raman散射峰对应的Raman活性振动模式分别为:位于210 cm−1散射峰对应于面间平动模式T;位于326 cm−1散射峰对应于面间振动模式L;位于737 cm−1散射峰对应于面内对称弯曲模式ν4;位于1093 cm−1散射峰对应于面内对称拉伸模式ν1;位于1444 cm−1散射峰对应于面内反向对称拉伸模式ν3;位于1760 cm−1散射峰对应于面外弯曲模式2ν2.

图2 高压合成产物碳酸镁样品的Raman散射图谱Fig.2.Raman spectrumof as-synthesized MgCO3sample and its Raman activemodes T,L,ν1,2ν2,ν3and ν4.
3.3 碳酸镁P-T相图的研究以及对其高压合成原理的解释
为了解释利用高温高压固相反应法合成碳酸镁的原理,通过TG分析以及尝试0.5,1.0,1.5 GPa压力下不同的反应温度条件的实验,我们对二水草酸镁-碳酸镁在P-T相图的稳定性进行了定量研究.
首先,分别对原料二水草酸镁和高压合成的碳酸镁进行TG分析.
图3(a)中黑线为二水草酸镁和合成的碳酸镁的TG曲线,采用氩气气氛升温速度为5◦C/min,温度范围40—1000◦C,测试样品质量为20 mg.从TG图可以看出,二水草酸镁一共有两次明显的失重行为,第一次位于温度范围约200—250◦C,TG由起始100%变为76.1%,第二次位于温度范围约450—550◦C,TG由76.1%变为26.9%.为了精确分析二水草酸镁TG产物,原位红外检测结果如图3(b)所示,图中给出了波数-温度-红外吸收的关系.可以明显看出:温度在200—300◦C范围,在波数1500 cm−1以及 3500 cm−1附近出现了 H2O的红外吸收峰;温度在400—600◦C范围,在波数600,2200以及3700 cm−1附近明显可见CO2的红外吸收峰,而在波数2200 cm−1附近存在CO的红外吸收峰,位置和CO2的红外吸收峰有所重叠.
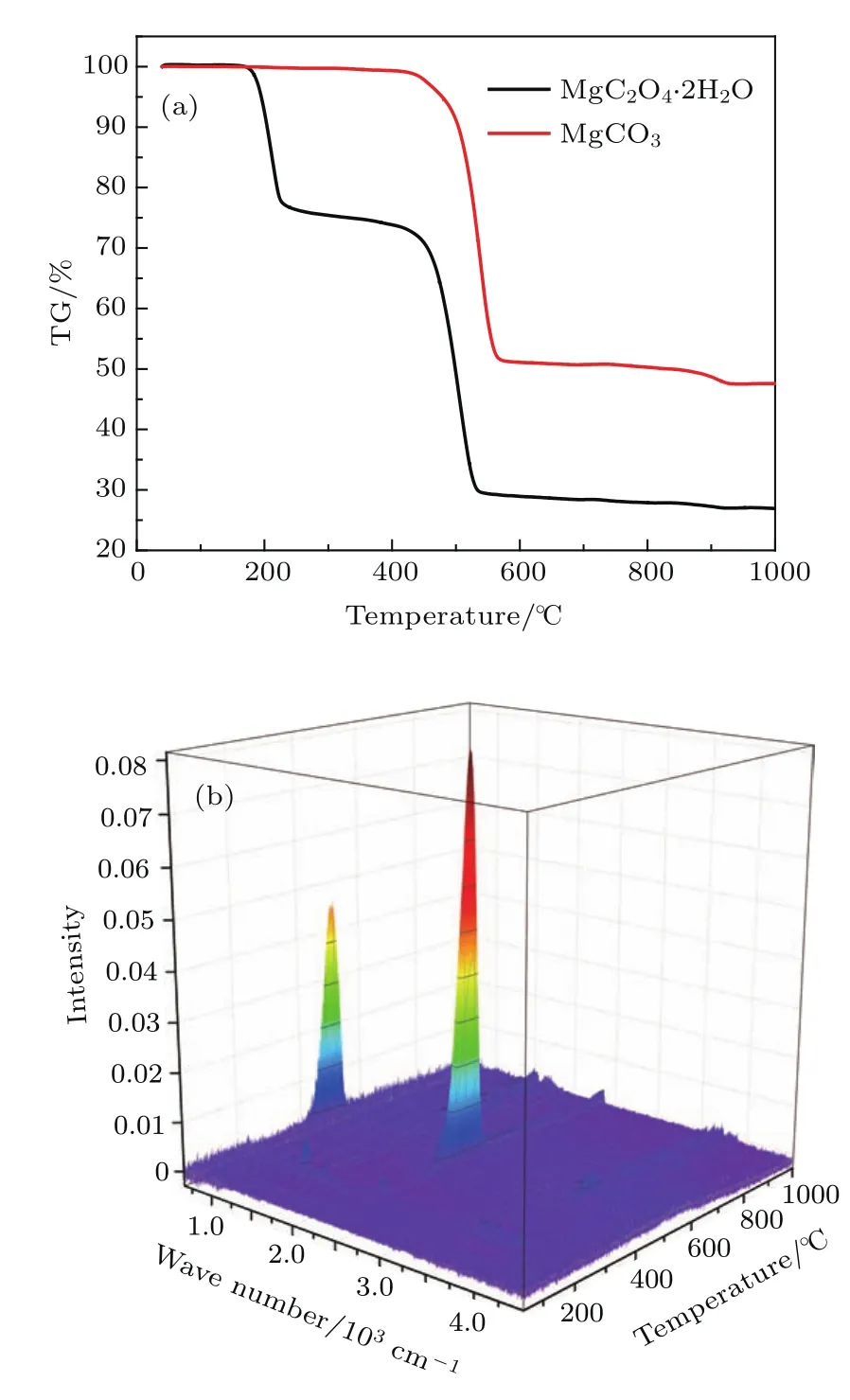
图3 (a)原料二水草酸镁和合成产物碳酸镁的TG曲线,实验条件为氩气气氛,升温速率为5◦C/min;(b)二水草酸镁TG分析过程中的红外光谱原位探测结果Fig.3.(a)TG analysesofMgC2O4.2H2Oand MgCO3at the heating rate of 5◦C/min in argon atmosphere;(b)in-situin frared spectrumof MgC2O4.2H2OTG analysis.
通过以上TG实验结果可以确定二水草酸镁在常压下的热分解过程.温度在200—250◦C范围内,发生脱水反应:MgC2O4.2H2O→MgC2O4+2H2O,TG曲线失重变为76.1%,失去两分子水的TG理论值为75.7%.温度在450—550◦C范围内,发生分解反应:MgC2O4→MgO+CO2+CO,TG曲线失重变为26.9%,失去一分子CO2和一分子CO的TG理论值为27.2%,实验值和理论值相符.
图3(a)中红线为高压合成的碳酸镁的TG曲线,在氩气气氛下升温速度为5◦C/min,温度范围40—1000◦C,测试样品质量为20 mg.从TG图可以看出,样品共有一次明显的失重行为,位于温度范围约450—550◦C,TG由起始100%变为47.9%.理论计算分解产物为MgO的TG理论值为47.8%,实验值和理论值符合得很好.
由二水草酸镁和碳酸镁的TG分析可知,二者都在温度约450—550◦C发生分解,二者分解温度区间几乎重叠.所以,在常压下,无法由二水草酸镁通过热分解直接得到碳酸镁.
高温高压条件是材料合成的一种极其有效的手段,尤其对于那些常压下极不稳定或者不存在的材料,可以尝试在高压下P-T相图中寻找其稳定相区,发现高压下的隐藏相(hidden phase),如果此隐藏相足够稳定能够淬灭到常压条件下,便成功地完成了该材料的高压合成.下面我们尝试在高压条件下合成碳酸镁.
一般情况下,高压条件能够使得某些材料的热稳定性得到很大的提高.在设定的压力条件下,碳酸镁的热分解温度得到大幅度提高,可以使碳酸镁和二水草酸镁分解温度区间发生分离.这使得在二水草酸镁-碳酸镁P-T相图中能够找到某一相区域,恰好能够使二水草酸镁相分解而碳酸镁相稳定.在这个相区域所对应的温度T和压力P条件下,便能够成功地合成碳酸镁.
表1为二水草酸镁在0.5,1.0和1.5 GPa压力和不同温度条件下的分解产物,图4(a)给出了对应的草酸镁-碳酸镁-氧化镁的P-T相图,其中,常压下二水草酸镁和碳酸镁均在450—550◦C分解,简单地设定二者的常压分解温度为450◦C,图中标出了二水草酸镁-碳酸镁的相稳定区域以及二者的相边界,红色虚线为碳酸镁-氧化镁示意的相边界,由于1.0和1.5 GPa时温度很高,实验上并未具体给出(高于800◦C,组装中的传压介质NaCl会熔化).由P-T相图可以明显看出,常压下碳酸镁的分解温度只有450◦C,0.5 GPa压力下,其在700◦C仍然稳定,而在1.0 GPa和1.5 GPa的压力下其直到800◦C都未分解.由于碳酸镁的热稳定性在高压下大幅度地提高,使得二水草酸镁的热分解(常压下脱水-分解两步过程)在高压下变成了一步,MgC2O4.2H2O → MgCO3+CO+2H2O,分解温度和脱水温度相当,约在200—300◦C之间,并且高压下不存在常压TG中看到的亚稳相MgC2O4,这个温度在高压下几乎没有变化(图4(a)中黑色虚线所示).进一步分析,在高压条件下,碳酸镁分解温度由450◦C提升到了至少700◦C,而草酸镁的分解温度由450◦C降低到了200—300◦C,由于高压使得二水草酸镁相和碳酸镁相原本重叠的热分解区域在P-T相图中发生了分离,导致在很宽的相区域内都能够使碳酸镁相稳定,并且压力越高,碳酸镁的相区域越宽,越利于碳酸镁相的稳定,这也解释了菱镁矿能够在下地幔如此之高的温压条件下保持稳定的原因.因此,我们可以通过高温高压下直接分解二水草酸镁的方法轻松地完成碳酸镁的制备,而这个过程是常压条件下难以完成的.

表1 不同高压合成条件下MgC2O4.2H2O的物相结果Tab le 1.Resu lts of assessment phase by high pressure synthesis fromMgC2O4.2H2Ounder various cond itions.

图4 (a)草酸镁-碳酸镁-氧化镁在0.5,1.0和1.5 GPa下的P-T相图;(b)在相同XRD测试条件下,1.0 GPa下的一组碳酸镁样品(104)主峰的相对峰强((104)峰强/(113)峰强)及其半高宽(FW HM)和合成温度的关系Fig.4.(a)MgC2O4.2H2O-MgCO3-MgOP-Tphase d iagramat high pressure of 0.5,1.0 and 1.5 GPa;(b)the(104)relative intensity(104)/(113)and its FW HMof a series of samples at various synthesis temperature under 1.0 GPa.
值得注意的是,在1.0 GPa压力条件和相同的反应时间下,反应温度为300,400,500,600,800◦C的一组样品都是MgCO3的纯相.在相同的XRD测试条件(0.02◦/step,采集时间2 s)下,比较这组样品的XRD图谱的主峰(104)的相对峰强((104)峰强/(113)峰强)和半高宽来判断样品的结晶度,如图4(b)所示.在相同的压力条件下,随着反应温度的升高,(104)主峰相对峰强增加,而其半高宽FWHM变小(在400◦C有变化,但不影响整体的规律).这说明合成温度越高,高压得到的碳酸镁样品的结晶程度越好,在800◦C下合成的碳酸镁样品的结晶程度最好.
3.4 碳酸镁形貌表征和晶粒度
在偏光显微镜下观察1.0 GPa,800◦C下合成的碳酸镁样品粉末形貌.图5(a)为碳酸镁样品在单偏光显微镜反光镜下的显微照片,图5(b)为碳酸镁样品在单偏光显微镜透光镜下的显微照片.在单偏光显微镜-反光镜下可以看出,样品平均颗粒为20µm,少数晶粒长大尺寸达到了200µm,呈板状菱形体的聚集体,有彩色光晕.在单偏光显微镜-透光镜下可以清晰地看到样品120◦的菱形解理面,这是天然菱镁矿常见的自然解理面 (101).由此可以判断,高压合成的碳酸镁样品结晶程度很好,这使得在单晶粒上表征Raman光谱以及微区电子探针的测试成为可能.另外,还可以通过控制温度压力以及反应时间、降温速率等实验条件,使晶粒进一步生长,这为将来尝试生长高质量的碳酸镁单晶提供了必要的实验手段和实验基础.

图5 高压合成的碳酸镁样品(a)在偏光显微镜-反光镜下的显微照片和(b)透光镜下的显微照片Fig.5.Micrographs of FeCO3under single polarizing microscopewith(a)refl ected light(b)and transmitted light.
3.5 用电子探针分析碳酸镁样品
为了解高压合成产物碳酸镁样品薄片的成分特征,我们用电子探针对其进行了成分分析.以MgO为镁含量的标准样,表2为样品中镁含量的电子探针分析结果,图6为样品的背散射电子图像以及探针探测位置.结果表明,在碳酸镁样品薄片的背散射电子图像中可以观察到所有晶粒的抛光面的灰度一致,说明样品的镁含量均一.不同区域共计的10个点的MgO质量百分数探针分析结果平均值为47.2%,计算可得样品成分为Mg0.99CO3,在误差范围内与理论相符.够很好地控制和重复.利用偏光显微镜观察样品形貌和晶粒度,清晰地看到了无水碳酸镁晶体的解理面,这些工作为将来高质量碳酸镁单晶的生长奠定了必要的实验基础.

表2 高压合成产物碳酸镁样品的电子探针分析结果Tab le 2. Resu lts of electron probe analysis of assynthesized MgCO3.

图6 高压合成产物碳酸镁的背散射电子图像Fig.6.The backscattered electronic of as-synthesized MgCO3.
4 结 论
我们利用高温高压固相反应法成功合成了无水碳酸镁,合成的样品具有纯度高、结晶程度好等特点,并且合成方法和条件控制比以往报道的方法相对简单.结合热重分析以及不同合成条件下的实验结果,找到了碳酸镁在P-T相图中的相稳定区域,合理地解释了高压合成无水碳酸镁的原理,并给出了0.5,1.0和1.5 GPa压力下的合成条件,P-T相图中碳酸镁的成相区域很宽,这使得实验操作能
[1]W ang A,Pasteris J D,Meyer HOA,Dele-Duboi ML 1996Earth P lanetary Sci.Lett.141 293
[2]Freitag F,Kleinebudde P 2003Eur.J.Pharmaceut.Sci.19 281
[3]Lou Z,Chen C,Chen Q 2005J.Phys.Chem.B109 10557
[4]Q ian J,McMu rray C E,Mukhopadhyay D K,W iggins J K,Vail MA,Bertagnolli KE 2012In t.J.Refractory Metals Hard Mater.31 71
[5]Surface J A,Skemer P,Hayes S E,Conradi MS 2013Environ.Sci.Technol.47 119
[6]de LeeuwN H,Parker S C 2000J.Chem.Phys.112 4326
[7]Morgan AB,Cogen J M,Opperman R S,Harris J D 2007Fire Mater.31 387
[8]RigoloM,W oodhams R T1992Polymer Eng.Sci.32 327
[9]Berg G W 1986Nature324 50
[10]Alt J C,Teagle D AH1999Geochim.Cosmochim.Acta63 1527
[11]Pal’yanov Y N,Sokol AG,Borzdov Y M,Khokhryakov AF,Sobolev N V 1999Nature400 417
[12]Isshiki M,Irifune T,Hirose K,OnoS,Ohishi Y,W atanuki T,Nishibori E,Takata M,Sakata M2004Nature427 60
[13]Oganov AR,OnoS,Ma Y,G lass C W,Garcia A2008Earth P lanetary Sci.Lett.273 38
[14]Lin J F,Struzhkin V V,Jacobsen S D,Hu MY,ChowP,Kung J,Liu H,MaoH,Hemley R J 2005Nature436 377
[15]Lavina B,Dera P,Downs R T,Prakapenka V,RiversM,Su tton S,N icol M2009Geophys.Res.Lett.36 L23306
[16]Lavina B,Dera P,Downs R T,Yang W,Sinogeikin S,Meng Y,Shen G,SchiferlD 2010Phys.Rev.B82 064110
[17]Chai L,Navrotsky A1993Con tribut.Mineral.Petrol.114 139
[18]Sandengen K,Jøsang L O,Baard K2008Ind.Eng.Chem.Res.47 1002
[19]X ing Z,HaoQ,Ju Z,Xu L,Qian Y 2010Mater.Lett.64 1401
[20]Herman R G,Bogdan C E,Sommer AJ,Simpson D R 1987Appl.Spectrosc.41 437
[21]Rividi N,van Zuilen M,Philippot P,Menez B,Godard G,Poidatz E 2010Astrobiology10 293
PACS:62.50.–p,81.10.Jt,61.05.cpDOI:10.7498/aps.66.036202
High pressu re synthesis of anhyd rousmagnesiumcarbonate(MgCO3)frommagnesiumoxalate d ihyd rate(MgC2O4.2H2O)and its characterization∗
Liang Wen1)Li Ze-Ming1)2)Wang Lu-Ying1)2)Chen Lin1)2)Li He-Ping1)†
1)(Institu te of Geochemistry,Chinese Academy of Sciences,Guiyang 550081,China)2)(University of Chinese Academy of Sciences,Beijing 100049,China)(Received 22 September 2016;revised manuscript received 11 October 2016)
Stimulated by the extensive application and research value,the study of anhydrousmagnesiumcarbonate(MgCO3)has been a sub ject of great concern recently,sothat a basic problemin designing a method of eff ectively synthesizing MgCO3is very worth considering.In previous studies,diff erentmethodswere reported tosynthesize MgCO3successfully but they still have some obvious defi ciencies.Themicro-particle sizes are toosmall tosatisfy the basic requirements of micro-analysis.Thus,it is needed toexplore the newmethods of artificially synthesizing MgCO3with the simple process and the high effi ciency.
By using magnesiumoxalate dihyd rate(MgC2O4.2H2O)as starting material,MgCO3sample is successfully synthesized by a solid reaction under high temperature and high pressure for the fi rst time in this work.The properties of as-synthesized sample are investigated by X-ray powder diff raction and Raman spectroscopy:neither of themshows any impurities existing in the sample.Significantly,the crystallinity quality is greatly improved in the terms of the maximumgrain sizes upto200micrometers,which could provide a base for MgCO3single crystal growth in the future.Moreover,compared with the results of previous studies,the reaction time of high pressure synthesis is controlled within 1 h sothat the effi ciency of the synthesis is greatly improved.
Based on thermogravimetric analyses and the results of high pressure experiment under the various pressures and temperatures,the P-Tphase diagrams of MgC2O4.2H2O-MgCO3-MgOat high pressures of 0.5,1.0 and 1.5 GPa are obtained,and in this case,it is reasonab le toexplain the principle of MgCO3synthesis under high pressure strictly.Fromthe P-Tdiagram,high pressure can greatly improve the thermal stability of material,and the decomposition temperature of MgCO3obviously increases with pressure increasing.However,due todecomposition temperature of MgCO3increasingmore quick ly than that of MgCO4.2H2O,the stable phase regions of MgC2O4.2H2Oand MgCO3are separated fromeach other,and hence,the corresponding temperature and pressure can be controlled todecompose the phase of MgC2O4.2H2Owhile stabilizing the phase of MgCO3soas toobtain MgCO3successfu lly.
Besides,by using polarizing microscope,the morphology of MgCO3sample as well as its crystal cleavage plane(10¯11)is observed clearly,and it is noted that as-synthesized MgCO3has good optical properties and high-quality crystallinity.The electron probing analysis for MgCO3thin section is performed toquantify the Mg content and the calculation indicates that the sample composition is Mg0.99CO3.
anhydrousmagnesiumcarbonate,high pressure synthesis
10.7498/aps.66.036202
∗中国科学院地球化学研究所135项目、国家重点研究发展计划(批准号:2016YFC0600100)和中国科学院重大科研装备研制项目(批准号:YZ200720)资助的课题.
†通信作者.E-mail:liheping@vip.gyig.ac.cn
*Project supported by the 135 Programof the Institu te of Geochemistry,Chinese Academy of Sciences,the National Key Research and Development Plan of China(Grant No.2016YFC0600100),and the Large-scale Scientific Apparatus Development Program,Chinese Academy of Sciences(G rant No.YZ200720).
†Corresponding author.E-mail:liheping@vip.gyig.ac.cn
猜你喜欢
世界科学技术-中医药现代化(2020年2期)2020-07-25
贡嘎山(2018年1期)2018-08-08
发明与创新·中学生(2018年1期)2018-02-02
小说月刊(2017年8期)2017-08-16
西安工程大学学报(2016年3期)2016-06-05
文物保护与考古科学(2016年4期)2016-05-17
中国卫生标准管理(2015年24期)2016-01-14
中国药业(2014年17期)2014-05-26
中国药业(2014年19期)2014-05-17
新疆农垦科技(2014年9期)2014-02-28
