
蛋白质复合物识别算法综述
2017-11-16 05:31汤希玮
长沙大学学报 2017年5期
汤希玮
(湖南第一师范学院信息科学与工程学院,湖南 长沙 410205)
蛋白质复合物识别算法综述
汤希玮
(湖南第一师范学院信息科学与工程学院,湖南 长沙 410205)
蛋白质复合物对理解细胞功能和细胞组织原则具有重要作用.高通量技术识别蛋白质复合物仍然相当不成熟.计算方法识别蛋白质复合物能在很大程度上弥补实验方法的不足.为能给生物学家提供有价值的参考意见,匹配统计量评估方法被用于比较八种典型的计算方法.评估结果显示,各种计算方法识别出的复合物均能较好地匹配真实的复合物.同时,不同的算法又具有各自不同的优势和不足,这给蛋白质复合物识别算法研究提供了上升空间.
蛋白质相互作用;蛋白质复合物;识别算法
蛋白质复合物由蛋白质-蛋白质相互作用网络中两两相互作用并紧密结合在一起的蛋白质组成.它能实现各种分子机制,进而执行大量生物功能,因此研究蛋白质复合物对理解细胞功能和细胞组织原则有着重要作用.如何从蛋白质-蛋白质相互作用网络中识别蛋白质复合物是生物信息学研究中的关键问题.早期的研究注重通过生物实验确定蛋白质复合物,但生物实验不仅耗费巨大,效率不高,而且测不准.与此同时,随着高通量技术的发展,大量蛋白质相互作用数据不断涌现[1, 2],这为通过计算方法识别蛋白质复合物打下了坚实的基础.因为蛋白质-蛋白质相互作用网络可以转化为一张图,图中的结点代表蛋白质,结点之间的边表示蛋白质之间存在的相互作用关系,那么识别蛋白质复合物的科学问题就可以转化为从图中识别密度子图的计算问题[3, 4].过去十年,出现了大量识别蛋白质复合物的计算方法.本文选择八种典型的计算方法分别运行于蛋白质相互作用网络,识别蛋白质复合物,然后采用被广泛接受的评价方法来确定这些复合物的生物学意义.一方面是为生物学家在选择计算工具时提供有价值的参考,另一方面也是为了蛋白质复合物识别算法研究提供新思路.
1 蛋白质复合物识别算法
在这一部分,我们按照算法发表的年代顺序逐一描述八种典型的计算方法.
算法一:分子复合物侦测算法(Molecular Complex Detection,MCODE).MCODE算法依据蛋白质-蛋白质相互作用网络中蛋白质之间的连通性确定蛋白质复合物,它能过滤非密度子图并识别出有重叠的密度子图(即蛋白质复合物)[5].该算法首先计算每个结点的局部邻居密度,并用密度值给该结点打分,然后,选择分值高的结点构建种子结点集合,最后从集合中的种子出发扩展成聚类.
算法二:马尔科夫聚类算法(Markov Clustering,MCL).
马尔科夫聚类算法在识别蛋白质复合物的过程中表现出了很强的健壮性[6].MCL算法在蛋白质-蛋白质相互作用网络中模拟随机游走,设计了膨胀和扩展两个操作,控制加权或非加权邻接矩阵.扩展操作将新概率值赋给所有的成对结点,膨胀操作通增加密度子图内部的游走概率并降低密度子图之间的游走概率来改变图中的游走方向.算法迭代执行这两个操作将网络分离为许多的密度子图.
算法三:边缘密度图聚类算法(Density-Periphery based graph Clustering ,DPClus).
Amin等人[7]提出的DPClus算法通过跟踪密度子图的边缘识别蛋白质复合物.DPClus首先基于边的两个顶点之间的共同邻居给每条边加权,进一步通过已加权的度,再给顶点加权.为了形成蛋白质复合物,DPClus先将具有最高权值的节点(种子节点)选为初始聚类,然后按照优先级,从邻居中选择顶点,迭代扩大聚类.邻居顶点的优先级由两个条件确定:(1)邻居顶点和聚类中每个顶点相连的边的权值的总和;(2)邻居顶点和聚类中每个顶点相连的边的总数.该算法不能用于节点数超过5000个的大规模蛋白质-蛋白质相互作用网络.
算法四:CFinder算法.
CFinder算法能识别蛋白质-蛋白质相互作用网络中的功能模块(或者蛋白质复合物)[8].CFinder算法将团的概念进一步扩展为K-团,K-团包括k个结点.两个相邻的K-团共享(k-1)个结点.该算法将所有相邻的K-团连接在一起构成更大的密度子图即K-团扩散聚类.这样,CFinder算法借助团扩散方法能够识别出重叠的密度子图(蛋白质复合物).
算法五:基于极大团的聚类算法(Clustering method based on Maximal Clique,CMC).
CMC是刘桂梅等人[9]提出的基于极大团识别蛋白质复合物的聚类算法.CMC首先用极大团挖掘算法获得所有极大团,然后基于文献[10]中可靠性度量给每个相互作用指派一个分值.那么每个团都能通过其加权密度得到一个分值.最后,CMC移除或者合并高度重叠的团,生产蛋白质复合物.如果两个团高度重叠,CMC或者合并两个团为一个更大的团,或者简单地删除分值更低的团.
算法六:核心扩展算法(COre-AttaCHment,COACH).
为了深刻理解蛋白质复合物的组织结构,吴敏等人[11]提出了一个称之为COACH的蛋白质复合物识别算法.该算法分两个阶段,在第一阶段,COACH从邻接图中定义核心顶点,然后识别蛋白质复合物的核心;在第二阶段,COACH将附加节点加入到核心中,形成有生物意义的结构.
算法七:速度与性能聚类算法(Speed and Performance In Clustering,SPICi).
SPICi是一种快速聚类算法,能够从大规模生物网络中识别蛋白质复合物[12].通过定义结点的密度和支持度两个概念,SPICi算法给网络中的结点加权并选择加权度最高的结点作为种子,以种子为初始聚类,从其邻居集合中选择密度和支持度足够大邻居结点加入聚类,迭代执行以上规则直到新加入结点的支持度和密度低于给定阈值为止,最后输出聚类(即蛋白质复合物)并从网络中删除聚类中的结点.
算法八:层次聚类算法(Hierarchical Clustering Protein Interaction Networks,HC-PINs).
HC-PINs是一种快速层次聚类算法[13].该算法提出了边聚类值的概念,用以描述边的邻居构成团的过程.边聚类值概念的提出,有效地避免了蛋白质-蛋白质网络中边聚类系数偏小的局限.又基于强模块和弱模块定义了更灵敏的λ模块.算法执行时,将网络中每个节点(蛋白质)初始化为聚类并计算网络中每条边的边聚类值,然后依据边聚类值降序排列所有边并不断将具有最大边聚类值的边添加到聚类中,直到λ模块出现.最后所有边都被分配到不同的λ模块中形成蛋白质复合物.
2 复合物评估方法
匹配统计量分析是一种常见的评价方法.它将算法识别出的复合物与已知的标准复合物进行比较和匹配.已知复合物是指由生物学家经过生物实验技术如串联亲和纯化等获得的复合物.一直以来, MIPS(Munich Information Center of Protein Sequences)数据库对酵母蛋白质复合物的分类被广泛用于产生蛋白质复合物参考集(即已知蛋白质复合物的标准集合).尽管这种分类是有价值的,但是,它已经不能适应该领域的最新发展现状了.所以,我们从CYC2008[14]下载了408个典型的复合物作为已知复合物,采用与文献[5]相同的得分方案以确定识别出的复合物怎样有效地匹配已知复合物.该方案采用重叠得分(OverlapScore,OS)具体描述复合物的匹配程度.OS的计算公式如下.

(1)
公式(1)中,i表示算法识别出的复合物与已知复合物之间共有的蛋白质数,a表示算法识别出的复合物中蛋白质的数量,b是指已知复合物中蛋白质的数量.OS的值为0意味着算法识别出的复合物与已知复合物没有共同的蛋白质,二者完全不匹配;OS为1表示算法识别出的复合物与已知复合物完全重叠,是最佳匹配.
真阳性数(TP)是在OS大于某一阈值的条件下,算法识别出的复合物集合中至少能够匹配一个已知复合物的复合物数目,假阳性数(FP)指算法识别出的复合物总数减去真阳性数(TP).假阴性数(FN)表示不能被算法识别出的复合物所匹配的已知复合物数目.从而可以定义灵敏度(Sn)[5]为:

(2)
特异性(Sp)[5]为

(3)
由此可以定义二者的调和平均值(F-score).

(4)
基于以上公式,我们设计并实现了一个Perl程序,它能自动完成算法输出的所有蛋白质复合物的匹配分析并计算三个评价指标.
3 使用的生物数据集
我们使用来自DIP(Database of Interacting Proteins,http://dip.doe-mbi.ucla.edu/dip/Download.cgi?SM=7/)数据库中酿酒酵母的蛋白质相互作用网络作为算法评价分析的原始数据.该网络包含4950个节点(蛋白质)和21788条边(相互作用).之所以没有选择其他数据库或其他物种,是因为DIP数据库中的基因产物的相互作用都是通过实际的生物实验检测到的,相对而言,其假阳性比较小,数据可靠性较高.与其他物种相比,酿酒酵母的蛋白质-蛋白质相互作用网络数据是最全面的.
4 实验结果比较和分析
我们分别用MCODE、MCL、CFinder、DPClus、CMC、COACH、SPICi和HC-PINs在酿酒酵母的蛋白质-蛋白质相互作用网络上识别复合物.然后将各自得到的复合物与已知复合物进行匹配,从整体上评价算法识别蛋白质复合物的质量.由于生物学家主要关注计算方法所产生的处理结果的生物学意义,所以我们不比较这八种算法的时间复杂度和空间复杂度.各算法的控制参数值来自算法设计的推荐.
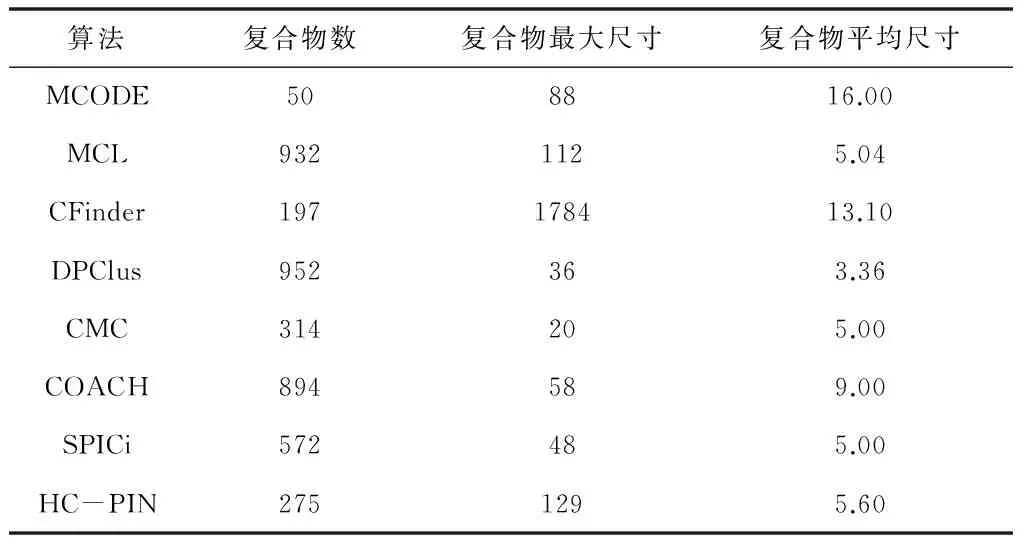
表1 八种算法识别出来的复合物的情况
表1显示了八种算法从蛋白质-蛋白质相互作用网络中识别出来的复合物的基本属性.其中,复合物最大尺寸是指包含最多蛋白质的复合物中蛋白质的数量,复合物平均尺寸是某个算法识别出的所有复合物包含的蛋白质的平均数.
从表1中可以看出,MCODE算法识别了50个蛋白质复合物,其中最大的复合物有88个蛋白质,所有复合物平均拥有16个蛋白质.MCODE算法依据图的局部密度扩充聚类,因此并不是蛋白质-蛋白质网络中的所有蛋白质都被指派到复合物中.与MCODE算法不同,MCL算法将蛋白质-蛋白质相互作用网络中的每个蛋白质都分配到相应的复合物中,它识别了932个复合物,仅次于DPClus算法的952个.CFinder算法识别了197个复合物,最大复合物覆盖了1784个蛋白质.MCL算法和SPICi算法都是将蛋白质-蛋白质相互作用网络分离为没有重叠的复合物,而其他算法采用一步一步的方式产生的复合物集合中包含了重叠的复合物.

图1 识别复合物与已知复合物在不同OS值上的匹配情况.
图1显示了匹配分析的综合比较结果.图1显示在OS的主要区间上,MCODE算法产生的复合物匹配上的已知复合物的数量最少,算法性能最差,相反COACH算法匹配的已知复合物的数量最多,表现最佳.文献[5]指出,当0.3≥OS≥0.2时,不具有生物学意义的蛋白质复合物已经被丢弃了.在这个范围内COACH算法匹配的蛋白质复合物数量最多,表现出了最佳的性能.
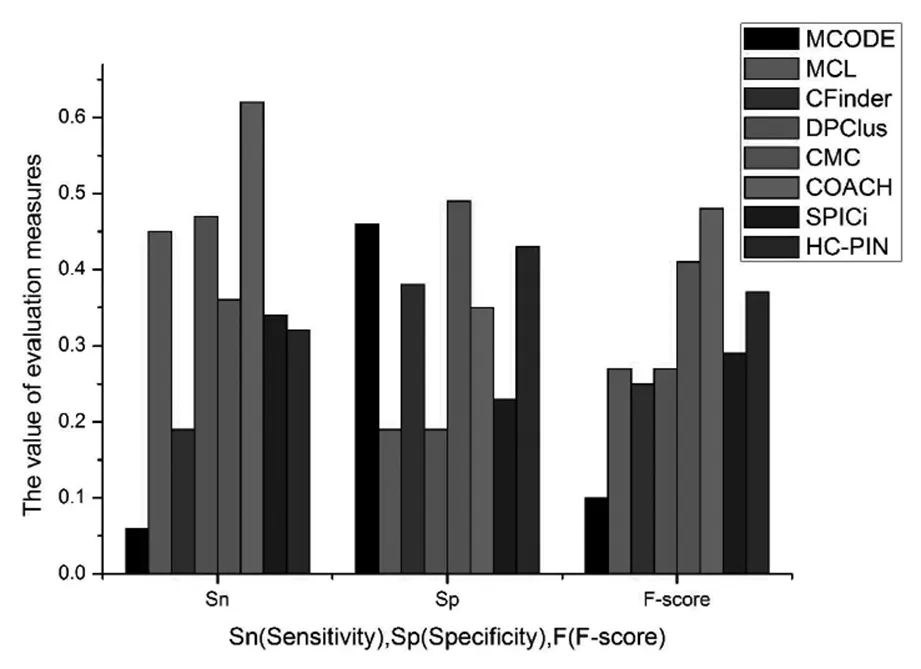
图2 基于三种评价指标的算法性能比较.
另外,我们的评估结果还显示 COACH算法产生了15个与已知复合物完全重叠的复合物,而HC-PINs产生了18个这样的复合物.
既然当0.3≥OS≥0.2时,大部分复合物都有生物意义,所以,我们取SO=0.2为阈值,计算了八种算法的灵敏度Sn、特异性Sp与调和平均值F-score.从图2可以看出,与其他算法相比,COACH、DPClus和MCL的灵敏度较高,这是由于它们识别出的复合物数量较多,则能够与已知复合物进行匹配的复合物数量也越多,所以灵敏度较高,SPICi的灵敏度较高也是这个原因.CFinder识别出了一个巨大的复合物,该复合物能够匹配的已知复合物数量应该是比较多的,导致其特异性较高,但其灵敏度并不高,可能的原因仍然是CFinder识别出的复合物数量偏少.CMC算法识别出的复合物数量远少于SPICi,而且最大复合物包含的蛋白质数也比SPICi对应的复合物包含的蛋白质数要少,但其灵敏度反而比SPICi高.这说明,CMC算法识别出的复合物能与更多的已知复合物匹配.与其他算法相比,CMC算法具有最高的特异性,这正说明其识别的蛋白质复合物质量高.通过以上分析,可以看出,单纯凭借灵敏度或特异性,都不足以说明算法的好坏.调和平均值F-score可以从一定程度上弥补了二者的不足,从数量和质量上,综合评价算法的性能.从F-score来看,COACH算法的性能最好,CMC算法次之.
匹配分析结果显示,尽管有些计算方法(如COACH、MCODE和HC-PIN)的性能在某些方面比其他计算方法要优异,但是,没有一种方法能占住绝对主导地位,全面超越其他算法.相反,实际上每种方法都各有优势以及不足,这也表明计算方法识别蛋白质复合物的研究仍有较大发展空间.
5 结束语
蛋白质复合物是执行细胞功能的关键分子实体.日益增加的大量蛋白质相互作用数据使得从蛋白质-蛋白质相互作用网络中识别蛋白质复合物成为可能.但也仅仅只是可能,因为高通量技术虽然能通过生物实验确定蛋白质-蛋白质之间的相互作用关系,但是并不能识别蛋白质复合物.幸运的是,计算方法正好能充当跨越鸿沟的桥梁.许多研究者对此展开了卓有成效的研究.本文选择的八种蛋白质复合物识别算法正是这一研究领域的典型代表.我们的实验结果表明各种算法虽然性能有高下之分,但是,识别出来的复合物都有一定的生物意义.这也暗示计算方法有助于生物学家继续寻找新的蛋白质复合物.
[1]Hamp T, Rost B. Evolutionary profiles improve protein-protein interaction prediction from sequence[J]. Bioinformatics, 2015, (12): 1945-1950.
[2]Li T, Wernersson R, Hansen R B, et al. A scored human protein-protein interaction network to catalyze genomic interpretation[J]. Nature Methods, 2017, (1): 61-64.
[3]You Z H, Li X, Chan K C. An improved sequence-based prediction protocol for protein-protein interactions using amino acids substitution matrix and rotation forest ensemble classifiers[J]. Neurocomputing, 2017, 228: 277-282.
[4]Wang L, You Z H, Chen X, et al. An ensemble approach for large-scale identification of protein-protein interactions using the alignments of multiple sequences[J]. Oncotarget, 2017,(3): 5149-5159.
[5]Bader G, Hogue C. An automated method for finding molecular complexes in large protein interaction networks[J]. BMC Bioinformatics, 2003, https://doi.org/10.1186/1471-2105-4-2.
[6]Van Dongen S. Graph Clustering by Flow Simulation[D]. Utrecht: PhD Thesis of Utrecht University, 2000.
[7]Altaf-Ul-Amin M, Shinbo Y, Mihara K, et al. Development and implementation of an algorithm for detection of protein complexes in large interaction networks[J]. BMC Bioinformatics, 2006,(1):1-13.
[8]Adamcsek B, Palla G, Farkas I J, et al.: locating cliques and overlapping modules in biological networks[J]. Bioinformatics, 2006, (8): 1021-1023
[9]Liu G M, Chua H N, Wong L. Complex discovery from weighted PPI networks[J]. Bioinformatics, 2009, (15):1891-1897.
[10]Liu G, Li J, Wong L. Assessing and Predicting Protein Interactions Using Both Local and Global Network Topological Metrics[J]. Genome Informatics, 2008, 21:138-149.
[11]Wu M, Li X L, Kwoh C K, et al. A core-attachment based method to detect protein complexes in PPI networks[J]. BMC Bioinformatics, 2009, (1):169.
[12]Peng J, Mona S. SPICi: A fast clustering algorithm for large biological networks[J]. Bioinformatics, 2010, (8): 1105-1111.
[13]Wang J, Li M, Chen J, et al. A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2011,(3): 607-620.
[14]Pu S, Wong J, Turner B, et al. Up-to-date catalogues of yeast protein complexes[J]. Nucleic Acids Res, 2009,(3): 825-831.
ASurveyonComputationalApproachesforIdentifyingProteinComplex
TANG Xiwei
(School of Information Science and Engineering, Hunan First Normal University, Changsha Hunan 410205, China)
Protein complexes are important for understanding principles of cellular organization and function. The high-throughput technologies for detecting protein complexes remain relatively immature. Computational approaches for identifying protein complexes are useful complements to the limited experimental methods. Eight state-of-the-art techniques for computational prediction of protein complexes are evaluated by matching method. Results show that the complexes identified by the eight computational methods match well with actual protein complexes. Meanwhile, different algorithms have their own comparative advantages and disadvantages, indicating that more research should be done in identifying protein complexes by computing methods.
protein-protein interaction; protein complex; identify algorithm
TP301.6
A
1008-4681(2017)05-0019-05
2017-09-07
国家自然科学基金面上项目“基于生物网络的人类疾病基因识别算法研究”(批准号:61472133).
汤希玮(1973— ),男,湖南常德人,湖南第一师范学院信息科学与工程学院副教授,博士.研究方向:生物信息学.
(责任编校:晴川)
猜你喜欢
中学生数理化·八年级物理人教版(2022年5期)2022-06-05
电子制作(2022年1期)2022-01-28
电子制作(2021年14期)2021-08-21
同济大学学报(自然科学版)(2019年2期)2019-04-02
计算机与数字工程(2019年3期)2019-03-26
西华师范大学学报(自然科学版)(2018年3期)2018-09-26
电子科技大学学报(2016年2期)2016-08-31
现代工业经济和信息化(2016年2期)2016-05-17
中国医学影像学杂志(2015年9期)2015-12-15
导航定位与授时(2014年2期)2014-04-27
