
摩擦改进剂减摩作用的分子模拟
2018-03-05 05:40李义雅段庆华代振宇
石油学报(石油加工) 2018年1期
李义雅, 龙 军, 赵 毅, 段庆华, 代振宇, 苏 朔
(中国石化 石油化工科学研究院, 北京 100083)
《中国制造2025》是我国实施制造强国战略第一个十年的行动纲领[1]。纲领中明确提出节能汽车对缓解我国能源与环境压力具有重要作用,低摩擦技术作为一种节能技术需要进一步研究改进。降低汽车使用中的摩擦可以通过完善发动机设计、改善车用材料、采用润滑节能技术等方法来实现。目前,改进润滑油的节能效果主要有适当降低润滑油的黏度和加入摩擦改进剂2种途径,通过降低表面间的摩擦系数,提高燃油经济性,减少能量损失。常用的摩擦改进剂一般由长直链烃基和极性端基组成,一般认为其作用机理为:极性基团通过物理或化学作用吸附在金属表面,烃基溶于油中,在金属表面定向排列形成稳定的分子层保护金属表面,且烃分子链具有一定柔顺性,从而发挥减摩作用[2]。目前多采用摩擦磨损试验机和现代分析技术研究摩擦改进剂的减摩性能,对不同摩擦改进剂的使用性能有了一定的认识[3-6]。
分子模拟为研究物质在原子分子水平的物理和化学性质提供了一种方法,可以更深入认识物质作用的本质,该方法在摩擦改进剂的作用机理研究方面已经有了较多应用。Tan等[7]采用从头计算方法,发现醇与羟基化金属表面的氢键作用比酯与羟基化金属表面的氢键作用强,而酯与裸露金属原子间的作用比醇与裸露金属原子间的作用强,因此醇和酯复配具有协同作用,能够发挥较好的减摩效果。Davidson等[8]采用分子动力学方法研究了摩擦改进剂的结构性能关系,结果表明,分子间氢键对形成的单分子层稳定性有重要作用,并且单甘油酯分子能够更有效地聚集形成更强的氢键;用密度曲线标示油溶性烃基与基础油的混合,烃基中扭转角的分布表明,长链烃基体系的结构与液体一致,而非晶体结构;用该方法预测的单、双、三甘油酯的减摩效率与实验测得的结果相一致。Xia等[9]采用量子化学研究了摩擦改进剂在Al表面的作用,根据计算结果,酯类比醇类的作用更强,与金属表面的作用位点是OC=O和OO-H。摩擦改进剂的润滑作用与分子中的官能团关系较大,前线轨道能级差等参数可以作为选择添加剂的理论依据。Ewen等[10]采用非平衡分子动力学研究了吸附在氧化铁表面的有机摩擦改进剂单分子层的原子结构和摩擦性质,结果表明,低、中、高覆盖范围的摩擦改进剂分子会分别形成类液体、无定形、类固体的单分子层结构,其摩擦系数也依次降低。
现有研究表明,摩擦改进剂分子与金属表面的物理和化学吸附作用是其发挥减摩作用的关键因素,从本质上认识这2种作用对开发新型摩擦改进剂有重要意义,而目前对摩擦改进剂分子这2种作用的系统研究还有所欠缺。笔者采用分子动力学和量子化学方法,以油酸和油酸酰胺为摩擦改进剂的模型化合物,以二十四烷基环己烷为基础油的模型化合物,研究2种摩擦改进剂分子与金属Fe表面的物理吸附和化学吸附作用本质,分析了温度对摩擦改进剂作用的影响。
1 分子模拟方法
1.1 计算模型和参数
选择油酸和油酸酰胺2种广泛应用的摩擦改进剂为模型化合物,其分子结构如图1的(1)和(2)所示。摩擦改进剂是在工况比较苛刻的条件下润滑油基础油表面膜难以满足润滑作用需求时发挥减摩作用的。为说明摩擦改进剂的作用机理,有必要比较摩擦改进剂与基础油发挥作用的本质不同。基础油的理想组分是少环长侧链的烃类和少分支的异构烷烃,其碳数分布为C20~ C40,选择二十四烷基环己烷为基础油的模型化合物,其分子结构如图1的(3)所示。
计算过程均采用Materials Studio 8.0软件,使用Forcite模块用分子动力学方法研究物理作用,使用DMol3模块用量子化学方法研究化学作用。分子动力学方法中,力场选择COMPASSⅡ,静电作用和范德华作用的非键截断均采用Ewald算法,计算的时间步长均为1×10-15s,有关压力和温度的控制函数分别采用Berendsen和Nose。量子化学采用基于广义梯度近似(GGA)的PBE泛函方法,k-point 选择(2×2×1),在DND基组水平上进行全电子计算。能量、受力和位移的收敛标准分别为5.25×10-2kJ/mol、105.02 kJ/(mol·nm)和5×10-4nm,自洽场(SCF)迭代收敛的阈值设为2.63×10-2kJ/mol。
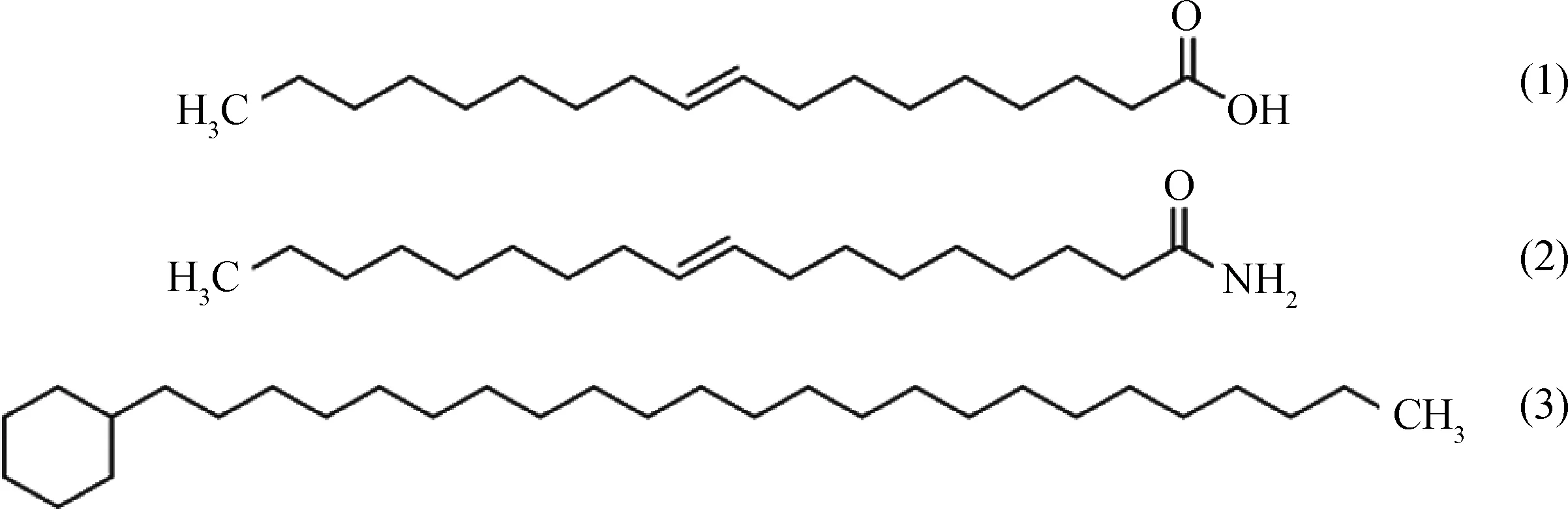
图1 模型化合物的分子结构Fig.1 Molecular structure of model compounds (1) Oleic acid; (2) Oleamide; (3) Tetracosyl cyclohexane (Cycloalkane)
由于金属Fe中Fe(110)表面是生长速率最慢的表面,在Fe晶体中所占比例最高,选择Fe(110)表面为金属表面。分子动力学的计算速率较快,选择较大的Fe(110)表面,构建表面体系的大小为2.9789 nm×2.9789 nm×1.2245 nm,厚度为7层。量子化学的计算效率有限,且化学吸附的作用位点主要是极性基团,C原子数大于3时,极性端基的电荷不再改变。因此为提高计算效率,用含有4个C原子及极性基团的简化分子结构进行量子化学计算,其结构如图2的(1)和(2)所示;选择戊烷为基础油分子的简化结构,如图2(3)所示。同时选择较小的Fe(110)表面,构建表面体系的大小为0.9930 nm×0.9930 nm×0.6081 nm,厚度为4层。

图2 量子化学计算的分子结构
Fig.2 Quantum chemical computational molecular structure (1) Butyric acid; (2) Butyramide; (3) Pentane
1.2 模拟过程
1.2.1 分子动力学计算过程
先用几何优化和分子动力学得到如图1所示的3种分子结构的最低能量构象,以油酸的最低能量构象为计算模型,用Amorphous Cell建立含有50个分子、密度为0.4 g/cm3的无定形晶胞。采用等温等压系综(NPT),对该晶胞进行退火及动力学平衡计算,平衡后统计计算1.01×105Pa、25℃下的密度,得到油酸在该条件下的密度为0.8755 g/cm3,实验值为0.8910 g/cm3,两者相近,说明选择的参数合理。用同样的方法,计算油酸酰胺和二十四烷基环己烷的密度分别为0.8735和0.8288 g/cm3。
采用Build Layers工具,以之前构建2.9789 nm×2.9789 nm×1.2245 nm大小的Fe(110)表面为金属表面层,搭建含有上、下2个Fe表面,中间有2.5 nm 真空层的层状模型。在该模型上添加Connolly表面,用Amorphous Cell的Packing方法,按照之前计算的密度,在真空层分别添加油酸、油酸酰胺和二十四烷基环己烷分子。为避免2个周期性结构之间的作用,在Fe(110)的上表面添加3 nm 的真空层,以油酸为例,最终搭建的润滑模型如图3所示。
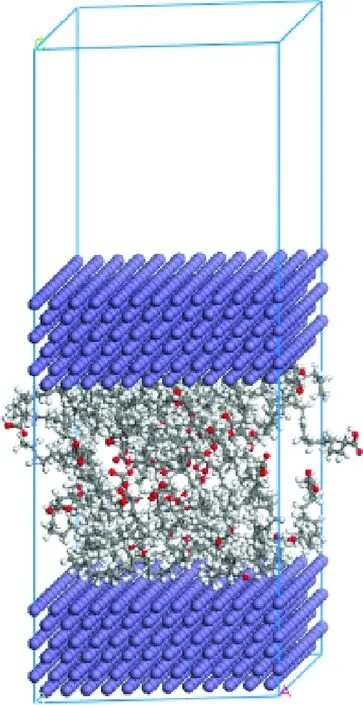
图3 分子动力学计算的润滑模型Fig.3 The lubrication model of molecular dynamics calculations
3种做动力学计算的模型分别在25、75、100、150、200、250、300、350、400℃条件下,用正则系综(NVT)对分子层作2×10-10s的动力学弛豫,使分子层在Fe(110)表面间的分布达到平衡状态。然后用受限剪切(Confined Shear)方法模拟剪切运动,上、下2个Fe(110)表面为限定层,沿x轴方向相对运动,中间的分子层在不同温度下作剪切运动,总模拟时间为1.5×10-9s。剪切运动之后,计算分子与Fe(110)表面的吸附能,以及分子之间的内聚能。
不同结构分子与Fe(110)表面的吸附能可通过公式(1)计算,分子的内聚能用Forcite模块的Cohesive Energy Density方法计算,其分子的内聚能由公式(2)得到。
Eads=Etot,1-(Emol+Esur)
(1)
Ecoh=Etot,2-Ein
(2)
式(1)、(2)中,Eads为分子与Fe(110)表面的吸附能,Etot,1为分子与Fe(110)表面作用后的总能量,Emol为无Fe(110)表面时分子的能量,Esur为无分子时上、下2个Fe(110)表面的能量,Ecoh为分子的内聚能,Etot,2为单独计算分子内聚能密度时得到的分子总能量,Ein为所有分子的内能,单位均为kJ/mol。
1.2.2 量子化学计算过程
为避免上、下2个周期性结构的分子有相互作用,在构建的较小Fe(110)表面添加2 nm的真空层。固定下面2层Fe原子,用DMol3优化含真空层的Fe(110)表面。用Forcite模块对图2所示3种简化的分子进行动力学平衡,再用DMol3优化确定分子的最低能量构象。将优化后的分子放到Fe(110)表面上,用几何优化确定分子与Fe(110)表面作用后的最低能量构象。
优化后,用CASTEP模块的Population Analysis分析有机分子中原子与Fe原子的电子重合百分率,用键级的值表示,用公式(1)计算分子与Fe(110)表面的吸附能。计算吸附过程的反应热(ΔE),判断反应过程中的热量变化。计算吸附前后分子中极性基团上的电荷分布,分析分子与Fe(110)表面作用过程中的电荷转移情况。
2 结果与讨论
2.1 摩擦改进剂的物理作用
2.1.1 摩擦改进剂的物理作用本质
摩擦改进剂可以通过物理或化学作用吸附在金属表面,形成表面吸附膜,发挥减摩作用。分子是否能够形成稳定的吸附膜与2种作用能相关,一是分子与金属表面的吸附能,另一种是分子的内聚能。用分子动力学方法研究摩擦改进剂的物理作用,计算剪切运动后油酸、油酸酰胺和环烷烃分子在25℃下与Fe(110)表面的吸附能及分子内聚能,并分析其能量组成,结果如表1所示。吸附及分子间的聚集过程都是放热过程,因此吸附能和内聚能均为负值,负值越小说明吸附和分子间聚集越容易发生,即其绝对值越大吸附能和内聚能也越大。由表1可以看出,25℃条件下3种分子层与Fe(110)表面的吸附能均远大于分子内聚能,初步说明在Fe(110)表面和分子层中分子对油酸、油酸酰胺和环烷烃分子均有吸引作用时,3种分子均可以克服分子层内分子的吸引作用,在Fe(110)表面的吸引作用下,运动扩散到Fe(110)表面并稳定吸附在表面上,并且吸附在表面的分子也较难由分子层内分子的引力使其脱附。同时分子的内聚能使吸附在表面的分子层具有一定强度,能够形成稳定的吸附膜,从而有效发挥减摩作用。可以得出,在25℃条件下油酸和油酸酰胺的2种作用能均比环烷烃分子的大,说明该温度下油酸和油酸酰胺分子形成表面吸附膜的稳定性比环烷烃吸附膜强。

表1 25℃条件下模型化合物的吸附能(Eads)及内聚能(Ecoh)Table 1 The adsorption energy (Eads) and cohesive energy (Ecoh) of model compounds at 25℃
EvdW—The van der Waals interaction energy among molecules;Eele—The electrostatic effect energy among molecules
详细分析2种作用能的能量组成,油酸、油酸酰胺和环烷烃分子与Fe(110)表面的吸附能均主要由范德华作用组成,基本没有静电作用,且油酸和油酸酰胺吸附能中的范德华作用均比环烷烃分子的大。油酸和油酸酰胺分子的内聚能由范德华作用和静电作用组成,主要是范德华作用;而环烷烃分子的内聚能基本只由范德华作用组成。使用氢键统计工具分析分子间的氢键作用,得到油酸和油酸酰胺分子间的氢键个数分别为26和31,氢键的平均长度为0.192 nm和0.210 nm,环烷烃分子间没有氢键。由计算结果可知,油酸和油酸酰胺的内聚能包括范德华作用、静电作用和氢键作用,范德华作用占主要部分;环烷烃的内聚能只包括范德华作用。比较各能量组成的大小可知,油酸和油酸酰胺的范德华作用与环烷烃相比差别不大,没有明显较高的优势;但是油酸和油酸酰胺分子的静电作用比环烷烃大,从而使油酸和油酸酰胺的内聚能比环烷烃的大。
为研究油酸和油酸酰胺的吸附能和内聚能均比环烷烃大的原因,分析油酸、油酸酰胺和环烷烃的电子密度分布,如图4所示,图中不同颜色代表不同的电子密度。由图4可以看到,油酸和油酸酰胺的电子分布不平均,OC=O上的电子密度较大;环烷烃的电子分布较平均,电子没有明显的聚集。说明油酸和油酸酰胺的正负电荷中心不重合,因此分子的极性较大;并且计算得到油酸、油酸酰胺和环烷烃的偶极矩分别为4.198、3.533和0.038 D,也可说明油酸和油酸酰胺的极性较大,环烷烃的极性较小。分子与Fe(110)表面作用过程中,Fe(110)表面为电中性,因此3种分子与Fe表面的吸附能均主要由范德华作用组成。而Fe(110)表面为固体,3种分子与其作用过程中会充分占据作用位点,表面一定,则3种分子层与Fe表面的作用位点也基本一致,由于油酸和油酸酰胺的极性较大,因此两者作用过程中的范德华作用较大,导致油酸和油酸酰胺的吸附能比环烷烃的大。对于分子间的范德华作用,虽然油酸和油酸酰胺的极性较大,但是环烷烃分子的碳数较多,原子排列紧密,使分子间的范德华作用差别不大。同时由于油酸和油酸酰胺的正负电荷中心不重合,使分子间具有静电作用;2种分子中的O—H和N—H键使分子间又具有氢键作用。因此油酸和油酸酰胺分子层内分子间的静电作用和氢键作用使它们的内聚能比环烷烃的大。
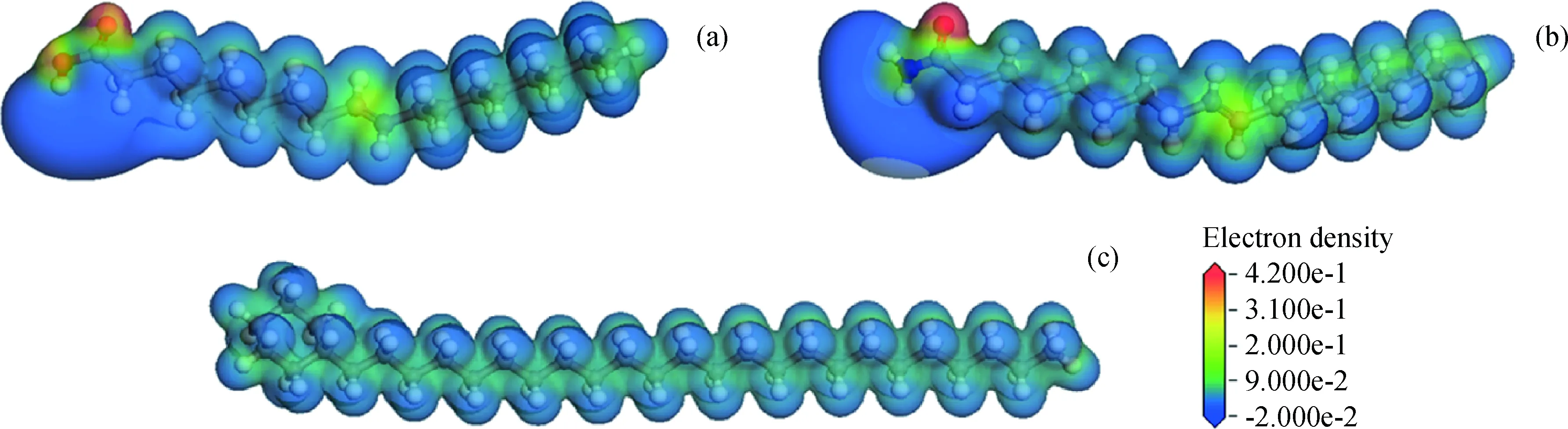
图4 模型分子的电子密度图Fig.4 The electron density maps of model molecules (a) Oleic acid; (b) Oleamide; (c) Cycloalkane
根据该计算结果可以得到,在25℃条件下润滑油组分形成稳定物理吸附膜的一个重要条件,即分子与Fe(110)表面的吸附能要远大于分子的内聚能,同时分子的内聚能使吸附膜具有一定强度。摩擦改进剂分子比基础油分子形成的吸附膜更加稳定的原因是,摩擦改进剂分子的电子分布不均,使其极性大,因此分子与Fe(110)表面作用的范德华作用能更大;且分子之间除了含有范德华作用,还有静电作用和氢键作用,从而使它们的吸附能和内聚能均较大,吸附膜也更稳定。由此推测,若要改进设计摩擦改进剂分子,本质是改变分子与Fe(110)表面的范德华作用,以及分子间的范德华作用、静电作用和氢键作用。
2.1.2 温度对摩擦改进剂物理作用的影响
温度是影响润滑油作用的一个重要条件,在较高的温度下,润滑油形成的润滑油膜失效,失去减摩抗磨作用。液体润滑油膜包括基础油和摩擦改进剂形成的吸附膜,研究温度如何影响润滑油膜的稳定性,对解决高温下如何改善膜的稳定性有重要作用。温度对油酸、油酸酰胺、环烷烃的吸附能和内聚能影响如图5所示。由图5可以看出,在不同温度下,3种分子与Fe(110)表面的吸附能均远大于分子的内聚能,分子可以克服分子层内分子间引力的阻碍扩散并吸附在Fe(110)表面,这是润滑油组分形成表面保护膜的一个重要条件。并且分子的内聚能使其具有一定强度,形成稳定的吸附膜,温度升高不会改变形成表面吸附膜的这2个条件。并且不同温度条件下,油酸和油酸酰胺的2种作用能均比环烷烃的大,所以油酸和油酸酰胺可以形成更加稳定的吸附膜,在更高的温度下发挥减摩作用。
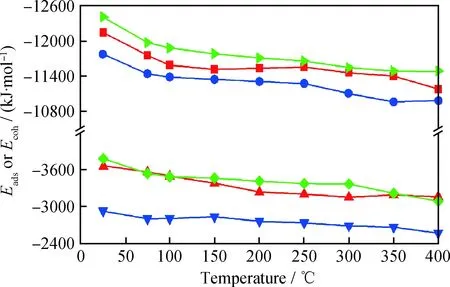
图5 温度对分子内聚能(Ecoh)和吸附能(Eads)的影响Fig.5 The influence of temperature on Ecoh and Eads of model molecules Eads (Oleic acid); Ecoh (Oleic acid); Eads (Oleamide); Ecoh (Oleamide); Eads (Cycloalkane); Ecoh (Cycloalkane)
由图5可知,随着温度升高,油酸、油酸酰胺和环烷烃的吸附能和内聚能均降低。分子形成吸附膜时,分子之间以及分子与Fe(110)表面间的作用能使吸附膜稳定存在,同时分子自身的动能会破坏这2种作用,影响吸附膜的稳定性。分析温度升高时3种分子动能的变化,结果如图6所示。由图6可以看出,随着温度升高,油酸、油酸酰胺和环烷烃的动能均增加,从而使吸附能和内聚能降低,吸附膜的稳定性降低,高温环境下吸附膜失效。同时可以发现,在同一温度下,油酸和油酸酰胺的动能均比环烷烃的动能小,这也导致油酸和油酸酰胺形成的吸附膜稳定性比环烷烃的好,能够在更高温度下发挥减摩作用。由上述分析可知,高温环境可以通过降低分子与Fe(110)表面的吸附能和分子层的内聚能两方面,降低润滑油膜的稳定性,从而使减摩作用失效。也可以说明,在高温环境下,可以通过提高润滑油膜与金属表面的吸附能以及分子的内聚能来增强吸附膜的稳定性,发挥减摩抗磨作用。
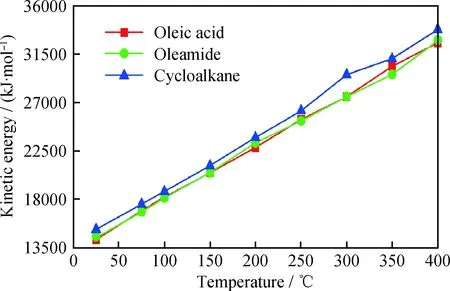
图6 温度对分子动能的影响Fig.6 The influence of temperature on kinetic energy of model molecules
2.2 摩擦改进剂的化学作用
除了物理作用,摩擦改进剂的极性基团还可以与金属表面发生化学作用。量子化学可以分析体系中电子的转移情况,采用基于密度泛函的量子化学方法研究化学作用,为提高计算效率,选择丁酸、丁酰胺和戊烷为模型化合物。作用过程中的吸附热(ΔE)和化学吸附能(Eads)如表2所示。由表2可以得到,丁酸和丁酰胺与Fe(110)表面的吸附能为负值,说明2个分子可以与Fe(110)表面发生吸附作用,且吸附能的绝对值较大,说明吸附作用较强;而戊烷与Fe(110)表面的吸附能为正值,说明戊烷与Fe(110)表面没有发生吸附作用。丁酸和丁酰胺与Fe(110)表面吸附过程的反应热均为负值,说明优化后产物的能量比反应物的能量低,该过程可以发生,即丁酸和丁酰胺可以与Fe(110)表面发生作用。

表2 量子化学计算后的反应热(ΔE)和吸附能(Eads)Table 2 The ΔE and Eads after quantum chemical calculations
为进一步研究化学作用过程,分析了吸附过程中丁酸和丁酰胺中杂原子与Fe(110)表面的距离变化,结果如图7所示。由图7可以看到,吸附后,丁酸中的OC=O与Fe(110)表面的距离减小,稳定时的距离较小,为0.2091 nm;而稳定时OO—H与Fe(110) 表面的距离较大,为0.3349 nm。丁酰胺中的OC=O与Fe(110)表面的距离减小,稳定时的距离较小,为0.2040 nm;稳定时NN—H与Fe(110)表面的距离较大,为0.3308 nm。吸附作用后,丁酸和丁酰胺中的OC=O与Fe(110)表面的距离较小,可能会有化学作用。CASTEP模拟计算中,原子之间的电子云重合百分数可以用原子之间键的键级表示,若发生化学作用,则该值大于0,说明原子之间有电子云重合,即2个原子之间有化学键。分析丁酸和丁酰胺中Fe—OC=O及Fe—OO—H、Fe—ON—H键的键级,得到丁酸中Fe—OC=O的键级为0.440,Fe—OO—H的键级为0,丁酰胺中Fe—OC=O的键级为0.480,Fe—ON—H的键级为0。由此可以说明,在吸附过程中,丁酸和丁酰胺中的Fe—OC=O可以与Fe(110)表面的Fe原子成键,发生化学作用,则丁酸和丁酰胺与Fe(110)表面发生化学作用的位点均是OC=O。分析戊烷中原子与Fe(110)表面的键级,均为0,说明戊烷与Fe(110)表面的吸附主要是物理作用。丁酸、丁酸酰胺和戊烷在Fe(110) 表面吸附后的稳态构象如图8所示,由图8可以看到,丁酸和丁酰胺中的OC=O与Fe原子距离较近,形成化学键。
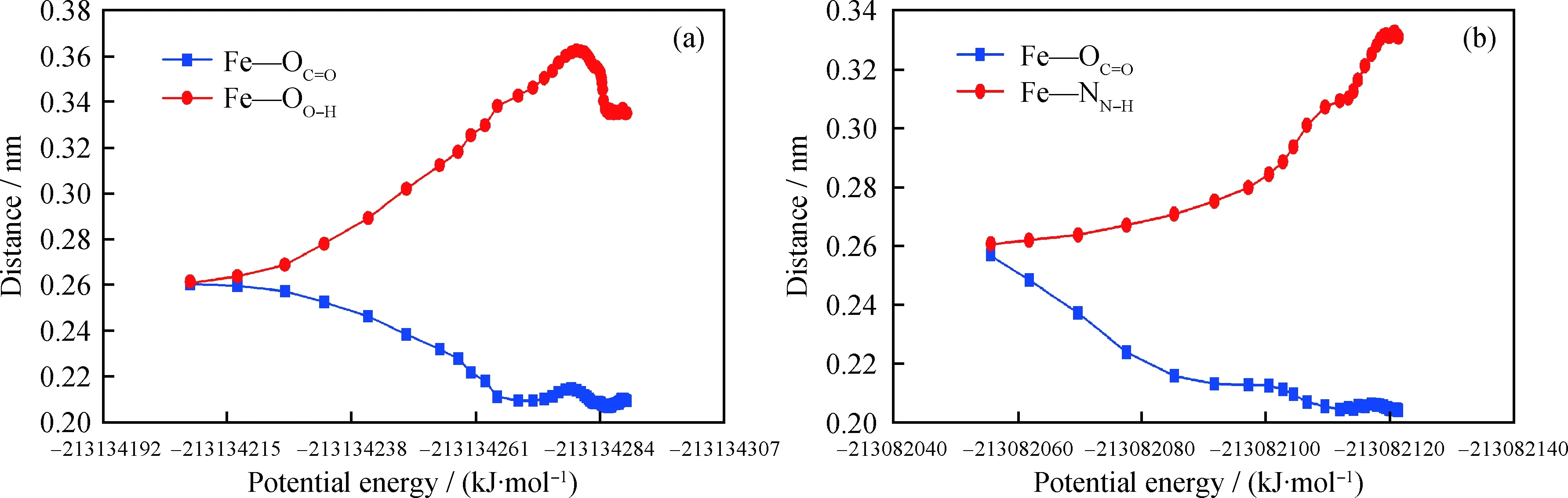
图7 吸附过程中丁酸、丁酸酰胺中杂原子与Fe(110)表面的距离Fig.7 The distance between heteroatom of butyric acid, butyramide and Fe(110) surfaces during the simulation (a) Butyric acid; (b) Butyramide
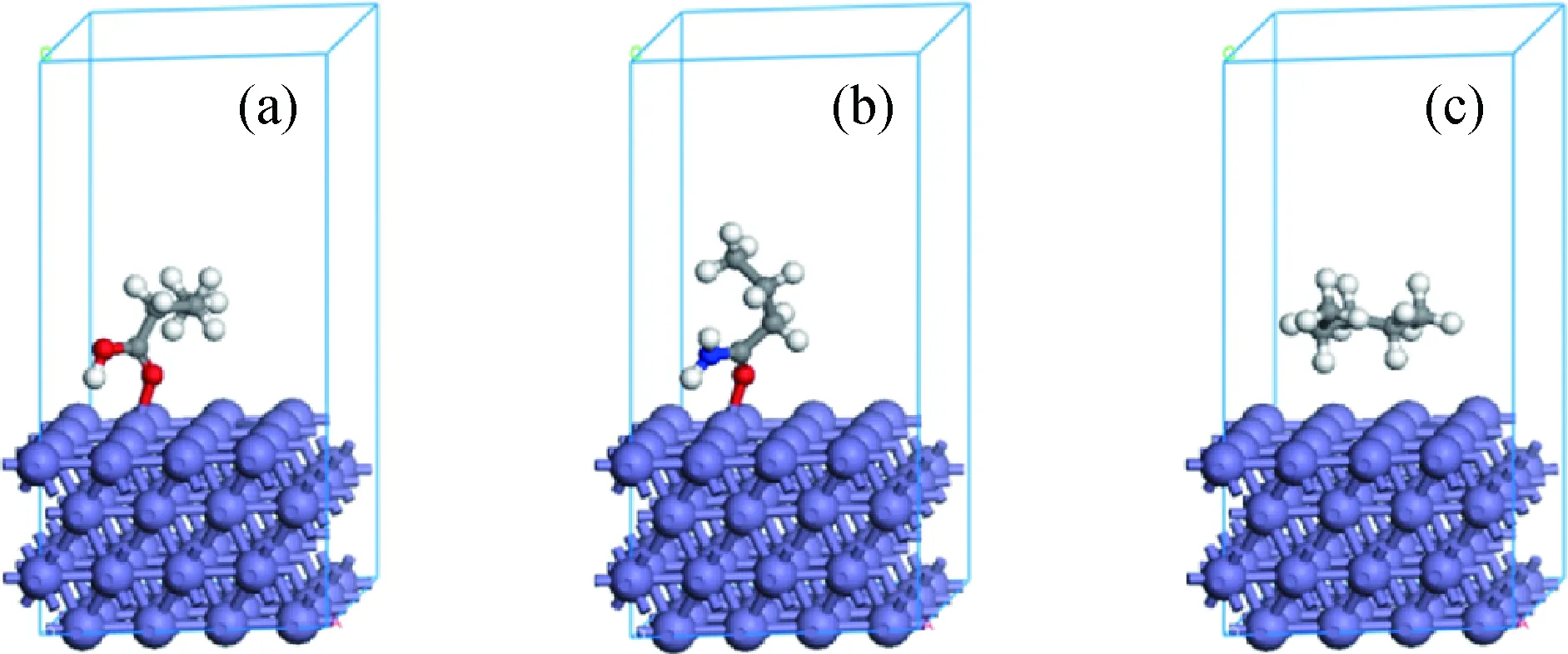
图8 丁酸、丁酰胺和戊烷在Fe(110)表面吸附后的稳态构象Fig.8 The stable conformations of butyric acid, butyramide and pentane adsorbing on the Fe(110) surfaces (a) Butyric acid; (b) Butyramide; (c) Pentane
计算分析化学吸附过程中作用位点的电荷分布,得到吸附之前丁酸和丁酰胺中OC=O的电荷分别为-0.261e 和-0.310e,原子与Fe(110)表面成键后的电荷分别为-0.140e和-0.172e;吸附后丁酸和丁酰胺中OC=O的电荷分别减少了0.121e和0.138e。由此可得,化学吸附过程中,丁酸和丁酰胺分子提供电子,Fe(110)表面接收电子,丁酸和丁酰胺转移的电荷量分别为0.121e和0.138e。根据之前分析的油酸和油酸酰胺电子密度分布,2个分子中均是OC=O上的电子密度最大,结合计算得到,分子通过向Fe(110)表面提供电子与其发生化学作用,则电子密度较大的位点更容易与Fe作用,因此可以说明电子密度较大的OC=O是最容易发生化学吸附的位点。同时可以得到,转移的电荷量与吸附能之间没有直接关系,丁酸转移的电荷比丁酰胺少,但丁酸与Fe(110)表面的吸附能比丁酰胺的大,转移电荷多的分子,化学吸附能不一定大。这可能是因为吸附是受多种因素影响的结果,化学吸附能的大小不只是与转移电荷量的大小有关。
综合物理作用与化学作用的研究,在Fe(110)表面与分子层内分子的共同吸引下,油酸和油酸酰胺分子能克服层内分子的引力运动扩散到Fe(110)表面,分子之间通过范德华作用、静电作用和氢键作用结合,在Fe(110)表面形成较稳定的物理吸附膜,发挥减摩作用。油酸和油酸酰胺中的官能团更加接近Fe(110)表面时,其中的OC=O通过向Fe表面转移电子与Fe原子成键,发生化学吸附。与环烷烃分子相比,由于油酸和油酸酰胺的极性较大,它们与Fe(110)表面的物理吸附能和分子内聚能均较大,并且除了物理吸附作用,它们的极性基团还可以与Fe表面发生化学吸附,因此油酸和油酸酰胺能够在更高的温度下发挥减摩作用。温度升高,分子的动能增加,使物理吸附能和内聚能均降低,也使形成的化学键强度减弱,因此在较高的温度下,油酸和油酸酰胺的减摩效果会减弱,需要稳定性更高的表面保护膜。
3 结 论
(1)油酸和油酸酰胺通过范德华作用与Fe(110)表面发生物理吸附,分子中OC=O通过向Fe表面提供电子与Fe(110)表面发生化学吸附,分子通过范德华作用、静电作用和氢键作用结合,且吸附能远大于分子内聚能,使其能够形成稳定的吸附膜,发挥减摩作用。
(2)由于油酸和油酸酰胺的电子分布不均,油酸和油酸酰胺与Fe(110)表面的物理吸附能和分子内聚能均比环烷烃分子的大,它们与Fe(110)表面还有化学吸附作用;并且油酸和油酸酰胺的动能均比环烷烃的小,因此油酸和油酸酰胺可以形成更加稳定的吸附膜,与环烷烃相比,能够在更高温度条件下发挥减摩作用。
(3)高温使原子、分子的动能增加,化学键强度减弱。因此,随温度升高,油酸和油酸酰胺形成吸附膜的稳定性下降,减摩作用逐渐减弱。
[1] 《中国制造2025》重点领域技术创新绿皮书[Z].北京: 国家制造强国建设战略咨询委员会, 2015: 90-96.
[2] RUDNICK L R. 润滑油添加剂化学与应用[M].李华峰, 李春风, 赵立涛, 等译. 北京: 中国石化出版社, 2006: 141-142.
[3] 蒋书运. 几种油性剂和极压抗磨剂对T8钢/Al2O3摩擦磨损性能的影响[J].摩擦学学报, 2004, 24(1): 29-32.(JIANG Shuyun. Tribological properties of T8 steel against Al2O3ceramics under the lubrication of 500SN oil containing various additives[J].Tribology, 2004, 24(1): 29-32.)
[4] 韦淡平. 环保型多效水溶性抗磨剂和摩擦改进剂的研究[J].石油学报(石油加工), 2011, 27(增刊1): 34-44.(WEI Danping. Study on environment friendly, water-soluble, and multifunctional antiwear and friction reduction additives[J].Acta Petrolei Sinica (Petroleum Processing Section), 2011, 27(S1): 34-44.)
[5] 刘琼, 龙军, 武志强, 等. 摩擦改进剂烷基链特性对减摩性能的影响[J].石油学报(石油加工), 2014, 30(2): 189-193.(LIU Qiong, LONG Jun, WU Zhiqiang, et al. Effect of alkyl chain characteristic of friction modifier on friction-reducing[J].Acta Petrolei Sinica (Petroleum Processing Section), 2014, 30(2): 189-193.)
[6] CAMPEN S, GREEN J H, LAMB G D, et al. In situ study of model organic friction modifiers using liquid cell AFM: Self-assembly of octadecylamine[J].Tribology Letter, 2015, 58(3): 1-15.
[7] TAN Y Q, HUANG W J, WANG X Y. Molecular orbital indexes criteria for friction modifiers in boundary lubrication[J].Tribology International, 2002, 35(6): 381-384.
[8] DAVIDSON J E, HINCHLEY S L, HARRIS S G, et al. Molecular dynamics simulations to aid the rational design of organic friction modifiers[J].Journal of Molecular Graphics and Modelling, 2006, 25(4): 495-506.
[9] XIA L, SUN J L, ZENG Y F, et al. Research on the relationship between molecular activity of additives and lubricating performance of aluminum rolling oil[J].China Petroleum Processing and Petrochemical Technology, 2013, 15(3): 13-18.
[10] EWEN J P, GATTINONI C, MORGAN N, et al. Nonequilibrium molecular dynamics simulations of organic friction modifiers adsorbed on iron oxide surfaces[J].Langmuir, 2016, 32(18): 4450-4463.
猜你喜欢
石油炼制与化工(2022年6期)2022-06-21
科学技术创新(2022年36期)2022-02-13
科学技术创新(2022年36期)2022-02-01
新农业(2020年18期)2021-01-07
煤炭转化(2020年2期)2020-04-24
中国机械工程(2019年7期)2019-04-23
都市家教·上半月(2017年7期)2017-08-15
润滑油(2016年4期)2016-11-04
中国民族医药杂志(2016年4期)2016-05-09
润滑油(2015年3期)2015-08-08
