
抗信号识别颗粒抗体阳性肌病的临床、病理及治疗
2018-10-12 11:21谢旭芳吴晓牧
中风与神经疾病杂志 2018年9期
黄 刚, 周 超, 谢旭芳, 吴晓牧
抗信号识别颗粒(signal recognition particle,SRP)抗体阳性肌病是一种免疫介导性坏死性肌病 ( IMNM ),以血抗信号识别颗粒抗体阳性为特点,常为急性或亚急性起病,多为成人发病,其病因多样,发病机制复杂、病理具有特殊性,临床表现及治疗个体差别大,其典型的临床特征为近端肢体对称性的肌无力,以及血清肌酸激酶(CK)显著升高,对皮质类固醇治疗部分反应或不反应。抗信号识别颗粒抗体阳性肌病发生率[1]在美国患者中为4%至5%,欧洲患者为6.4%,日本患者为8.3%,甚至更高(13.0%),在现有的研究中[1]表明抗SRP抗体阳性肌病在亚洲的特发性肌炎患者中可能更常见。现收集我科2008年~2017年诊断的抗SRP抗体阳性肌病8例,结合患者的临床资料、肌肉病理及治疗情况进行分析总结,旨在给临床医师提供借鉴。
1 对象与方法
1.1 一般资料 8例患者均来自于2008年1月~2017年12月在江西省人民医院就诊并诊断为抗SRP抗体阳性肌病的患者,所有的患者均通过肌炎抗体系列检查及肌肉病理证实的。患者的肢体无力均使用MRC分级法进行分级;疾病复发定义为在排除可能的肿瘤或感染过程后,归因于疾病活性的临床症状的恢复和(或)血清中CPK水平的增加。
1.2 方法 8例患者均常规进行血清肌酸激酶(CK)、肿瘤标志物、肌炎抗体谱、风湿免疫系列等测定、心电图或心脏超声检查、肺部CT检查、双大腿肌肉磁共振检查、以及其他相关检查。
8例患者在治疗前均行了肌肉病理,活检部位为肌力中等减退的肌肉或以肌肉磁共振的结果选择活检部位,避开肌电图检查的部位。活检的肌肉为三角肌或股四头肌,肌肉取出后常规液氮异戊烷固定并进行冰冻切片,行HE、MGT、ORO、PAS、NADH染色,并在光镜下进行观察。
1.3 治疗 5例患者初次诊断后给予了激素联合甲氨蝶呤治疗,2例患者初次诊断后只给予了激素治疗,1例患者因为病情较重给予了激素联合丙种球蛋白治疗;3例患者复发后给予了激素联合甲氨蝶呤治疗;6例初次诊断的患者为静脉使用的激素为甲强龙 1 g/d ×5 d 后改为口服强的松1 mg/(kg·d),2例初次诊断患者及3例复发的患者是直接口服强的松 1 mg/(kg·d);1例初次诊断患者使用丙种球蛋白剂量0.4 g/(kg·d)×5 d。
2 结 果
2.1 临床特点 男性1例,女性7例,年龄17~66岁,平均年龄37.5岁,平均发病时间3.5(1.8~7.8) m,病程4 m~2 y,病程呈亚急性的6 例,呈慢性的2 例;没有患者有心脏,皮肤或神经系统受累的表现,有1例合并了间质性肺病;所有患者都有客观的四肢肌肉无力[上肢和(或)下肢],大部分为Ⅲ或Ⅳ级,没有发现有颈部肌肉受损的表现;所有患者中有3例未复发,5例复发。
2.2 实验室检查 8例患者的CK在初诊时为2171~8705 U/L(平均4888.6 U/L)之间(见表1);肌炎抗体谱检测表现为所有的患者SRP抗体均为强阳性(有5例患者为复发后行了肌炎抗体检测),2例患者同时伴有R0-52抗体阳性,其余的抗体为阴性(见表1);风湿免疫系列检查:5例患者血沉升高,1例患者类风湿因子及抗链球菌溶血素O升高,1例抗核抗体阳性;肺部CT:1例患者提示间质性肺炎,其余正常;心电图或心脏超声检查正常。
2.3 肌肉磁共振检查 7例患者行了大腿肌肉磁共振检查;7例患者的大腿磁共振的脂肪抑制序列(STIR)可见部分肌肉内有斑片状高信号改变,甚至有斑片状高信号融合,提示肌肉组织水肿;同时伴有1例患者在FLAIR序列可见部分肌肉有高信号改变,提示脂肪沉积(见图1)。
2.4 电生理检查 6例患者为肌源性损害,即MUAP时限变窄、波幅降低、多相波增多;1例患者为神经源性损害,即MUAP时限正常、波幅增高;1例患者肌源性损害伴周围神经损害。
2.5 病理检查 7例患者仅出现少量散在分布的变性坏死和再生肌纤维,1例患者为肌营养不良样的病理表现,为可见肌纤维肥大、萎缩及变性坏死和再生肌纤维,伴随结缔组织增生;7例患者病理表现中未见炎性细胞,1例可见少量的炎性细胞(见图1)。
2.6 疗效 3例激素联合甲氨蝶呤治疗的患者半年后门诊复诊症状体征基本恢复,肌酸激酶及抗SRP抗体正常。5例患者与首次发病后3 m~3 y复发(即激素减量或停药后),症状较轻,CK升高,抗SRP抗体检测阳性,并排外了可能的肿瘤或感染,给予了激素联合甲氨蝶呤治疗后,半年复诊体征基本恢复,肌酸激酶及抗SRP抗体正常。目前8位患者一直在服用强的松(10 mg/d)和甲氨蝶呤维持治疗(见表1)。

表1 8例抗SRP抗体阳性肌病患者的临床、实验室检查及治疗资料
注:为强阳性;*伴有间质性肺炎;★伴有R0-52抗体强阳性;MTX:甲氨蝶呤
3 讨 论
特发性炎症性肌病(idiopathic inflammatory myopathies,IIM)是一组病因不明,主要影响肢体近端肌肉的获得性自身免疫性肌病。起病形式可以表现为急性、亚急性及慢性起病,根据临床及肌肉组织病理学表现,特发性炎症性肌病分为多发性肌炎(PM)、皮肌炎(DM)、非特异性肌炎(NSM)、包涵体肌炎(IBM)和免疫介导的坏死性肌病(IMNM)5 种亚型。免疫介导坏死性肌病是特发性炎症性肌病的亚型,其病理组织学特点为可见肌纤维坏死和再生,而无或极少的炎性细胞浸润,是由Mccombs等于1947年首先描述的,于1963年由Krolikowska等提出坏死性肌病的概念,于1991年Emslie-Smith等对坏死性肌病的病理特点进行了详细的描述[2]。免疫介导坏死性肌病的病因包括自身抗体介导、药物诱导、副肿瘤和病毒感染等,抗SRP抗体的存在是其最常见的病因,又称抗SRP抗体阳性肌病,而且有研究表明抗SRP抗体阳性肌病在坏死性肌病中约占53%[3]。
抗SRP抗体阳性肌病是Reeves等[4]于1986年首次报道的,其描述了一位患有多发性肌炎的患者,其血清中检测到抗SRP抗体,以后许多国家都陆续报道了抗SRP抗体阳性肌病,特别是在日本。有研究表明约4%~6%的特发性炎性肌炎(IIM)患者中检测到SRP的抗体,2014年国内学者[1]研究了123例诊断为特发性炎性肌病患者中发现抗SRP抗体阳性肌病为16例(13.0%),女性占优势,从而表明我国该类疾病并不少见。根据抗SRP抗体阳性肌病的临床表现和肌肉病理学特征,许多学者认为这种肌病不同于典型的多发性肌炎,而属于坏死性肌病的范畴,但临床上常常被误诊为多发性肌炎。抗SRP抗体阳性肌病通常是急性或亚急性发病,典型的临床特征是多为成人发病,主要表现为躯干及肢体近端对称的肌无力,但是轻度的无力、肌痛和呼吸困难常常可能是该病的初始症状,而且间质性肺炎和吞咽困难是常常可能是其的伴随症状,血清肌酸激酶显著升高,以及对激素治疗效果不完全或不反应。近年来有儿童发病的病例,在儿童的肌病中约小于1%,在儿童和成人患者之间的肌肉病理学发现似乎没有明显的差异,但抗SRP抗体阳性肌病的儿科患者的肌肉萎缩和无力发展更迅速、更显著,复发的风险很高[5]。本组患者临床表现为女性比男性多,多为亚急性起病进行性加重,肢体对称性无力是以近端为主,程度较远端更重,血肌酸激酶明显升高,血的抗SRP抗体强阳性,与文献报道相类似,另外,国内外文献均未发现该病与其他系统损害(如心脏、皮肤、肺部等)有必然的联系,本组患者只有1例其他系统受累患者,也没有发现两者之间有明确的联系。
抗SRP抗体阳性肌病是由于患者体内的血清存在抗SRP抗体而被命名的,SRP是与内质网相关的蛋白质,是由6个多肽和7S RNA分子组成的复合物,在血清中能够检测到6个SRP亚基或7S RNA,其中54-kDa SRP亚基在该蛋白质中起着主要的生理作用,因为所有六种SRP亚基都能被抗SRP抗体识别,但是SRP54亚基被抗SRP抗体识别在该病中总是存在的,因此SRP54被认为是抗SRP抗体最常见的靶点[6,7]。抗SRP抗体在体外对肌细胞融合有损伤并可诱导肌纤维萎缩,其机制可能是通过促炎分子(il-6,TNF,ROS)的增加、同时使2种抗炎细胞因子(il-4和il-13)的降低,抑制了肌细胞融合及导致肌纤维直径减少[6]。有研究表明该病治疗前抗SRP抗体浓度与疾病严重程度无相关性,但是在治疗后抗SRP抗体滴度与CK水平和肌肉无力程度是高度相关,这些也提示了该抗体在坏死性肌病中致病作用的可能性,但是日本学者H Hanaoka等研究中发现日本抗SRP抗体阳性的结缔组织病患者中大约1/3的患者没有发生肌病[8],另外近年来也有其他的学者发现该抗体在其他自身免疫性疾病中被发现过,因此抗SRP抗体的特异性目前有争议的。
抗SRP抗体阳性肌病的肌肉病理表现[1]类似于免疫介导坏死性肌病,为坏死和再生肌纤维,无或少量的炎性细胞浸润,少部分严重的患者存在肌纤维肥大和结缔组织增生常常被误诊为肢带型肌营养不良。本组患者的肌肉病理与文献报道的相一致,主要表现为散在的坏死和再生的肌纤维,炎性细胞浸润不明显,但是其中有1例表现与其他的表现不一样,表现为肌纤维明显萎缩并伴有肥大的肌纤维,大量的坏死和再生的肌纤维,还可见少量的炎性细胞,曾经被怀疑为肌营养不良,后因血抗SRP抗体阳性而最后诊断为抗SRP抗体阳性肌病。因此肌肉病理怀疑为肌营养不良时要排外抗SRP抗体阳性肌病可能,建议行肌炎抗体检测以减少误诊。
Kao等[3,9]研究表明其研究对象中有三分之一的成年患者使用皮质类固醇联合免疫抑制剂治疗能够实现了较好疗效,并且指出使用免疫抑制剂能减少皮质类固醇的副作用。2006年Arlet等[10]对2例难治性抗SRP抗体阳性肌病患者应用强的松、血浆置换、利妥昔单抗联合治疗取得显著疗效后,2012年Savey 等[11]用血浆置换和利妥昔单抗治疗抗SRP抗体阳性肌病 也取得较好疗效。因此目前大多数学者认为该病的治疗要皮质类固醇、免疫抑制剂、血浆置换、免疫球蛋白等治疗对于该病均有疗效,并认为[3 ]皮质类固醇联合静脉注射免疫球蛋白或免疫抑制剂作为一线治疗,血浆置换抗SRP抗体为是二线疗法。但是激素治疗目前有争议,认为该病对激素的疗效反应表现为不完全或不反应。本组患者中有3例在初次确诊后即使用了激素联合甲氨蝶呤治疗,疗效较好,没有复发,而有4例患者在初次确诊后只给予了激素治疗,有一定的疗效,但在激素减量停药后再次复发,提示了该病对激素治疗的反应性不完全,与文献报道相似,因此该病的治疗尽早使用激素联合免疫抑制剂治疗,对该病的预后有着重要的作用。另外根据本组1例重症患者治疗的体会,重症患者建议早期可激素联合丙种球蛋白,然后再使用激素联合免疫抑制剂治疗,可以取得较好的疗效。另外有研究表明[10,12]抗SRP抗体滴度虽然不能反应疾病的严重程度,但是能够反应该病的疗效,因为随着治疗的有效抗SRP抗体滴度下降并转为阴性,因此抗SRP抗体阳性肌病疗效除了临床症状体征外抗SRP抗体的滴度及CK浓度也能判断疗效。本组患者随着治疗的有效CK及抗SRP抗体滴度下降恢复正常,但是复发患者的CK及抗SRP抗体滴度会再次升高。
综上,抗SRP抗体阳性肌病在我国的发病率不详,但是并不少见。该病的临床表现具有相当大的变异性,容易导致误诊,目前的诊断主要依靠血液中的抗SRP抗体检测及肌肉病理,由于该病对激素治疗的反应性不完全,易复发,而激素联合免疫抑制剂疗效较好,因此确诊是关键。通过对我科诊断的抗SRP抗体阳性肌病的资料分析并结合文献复习,旨在增加临床医师对该疾病的认识,提高诊断率,减少漏诊误诊,从而提高该疾病的治疗效果减少复发。
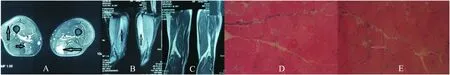
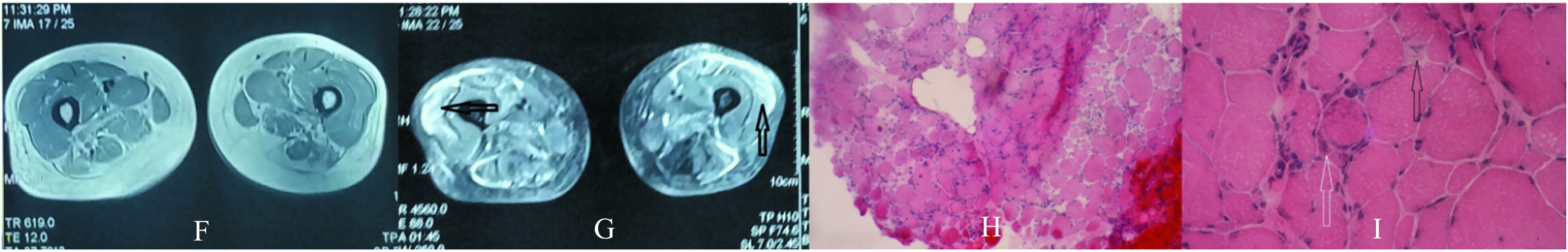
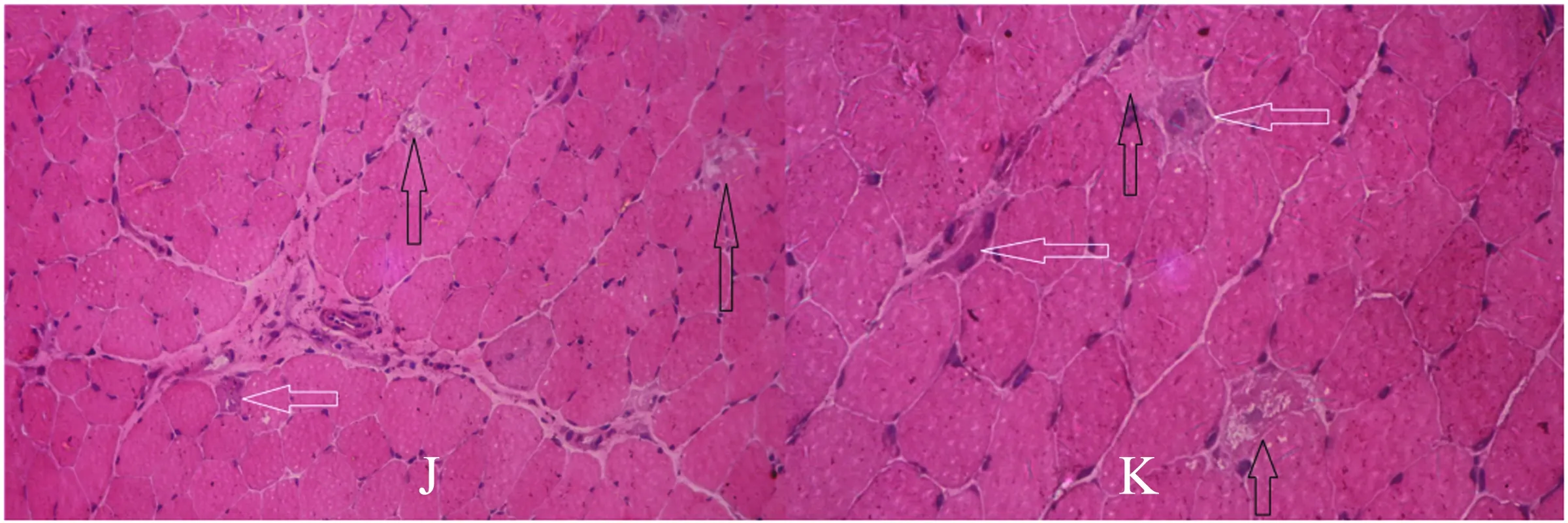
图1 A、B、C、D、E为例1患者;F、G、H、I为例3患者;J、K为例8患者;A、B、C、F、G为大腿磁共振检查(A、B、G为STIR序列;C、F为FLAIR序列):可见部分肌肉在STIR序列呈片状信号增高提示水肿(黑色箭头),肌容积减小、肌间隙增宽提示部分肌肉萎缩。肌肉病理(HE染色)(H×100,D、J×200,E、I、K×400):例1和例8患者可见少量的坏死肌纤维(黑色箭头)及再生的肌纤维(白色箭头),未见炎性细胞;例3患者可见大量的大小不等的圆型肌纤维、核集聚,坏死(黑色箭头)和再生的肌纤维(白色箭头),少量的炎性细胞
猜你喜欢
中国现代医生(2022年21期)2022-08-22
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
现代临床医学(2022年2期)2022-04-19
中国医院院长(2021年21期)2022-01-21
今日农业(2021年5期)2021-11-27
国际放射医学核医学杂志(2021年10期)2021-02-28
医学信息(2020年12期)2020-07-27
中外女性健康研究(2019年22期)2019-12-30
食品安全导刊(2018年36期)2018-05-25
