
气相色谱-质谱法测定水样中草甘膦和氨甲基膦酸的残留量
2019-01-23 02:14田应涛黎其万梅文泉耿慧春朱永芬
云南农业科技 2019年1期
田应涛,黎其万,梅文泉*,耿慧春,朱永芬
(1.屏边县和平镇农技站,云南屏边661202;2.云南省农业科学院质量标准与检测技术研究所,云南昆明650205;3.屏边县农业与科学技术局,云南屏边651200)
草甘膦(Glyphosate)又称镇草宁、农达(Roμndμp)、草干膦、膦甘酸,化学名为 N-(膦酰基甲基)甘氨酸,化学分子式C3H8NO5P,属于弱有机酸,是一种常用的水溶性除草剂,纯品为非挥发性白色固体,不溶于一般有机溶剂,且不可燃,常温贮存稳定[1]。草甘膦为内吸传导性强的广谱灭生性除草剂,其主要代谢产物为氨甲基膦酸,它们均呈弱酸性,极性高,水溶性强,不易溶于有机溶剂[2]。近年来,随着转基因抗草甘膦农作物增多,草甘膦用量逐年增加,已成为世界上应用最广、产量最大的农药品种,全球每年需草甘膦原粉40万t,占据世界农药销售额首位,且每年以20%左右速度递增[3~4]。在农业上大量使用的草甘膦除草剂,只有极少部分发挥了作用,多数残留在土壤或漂浮在大气中,通过降雨和径流的冲刷进入地表水或地下水,造成水体污染。随着世界各国草甘膦需求量和使用量的不断增加,人们对于草甘膦潜在的威胁更加关注,如长期饮用含有草甘膦农药的水,将会危及人体健康,并会造成肝肾、黏膜(消化道、皮肤)、神经、呼吸和心血管系统的损伤,严重者可危及生命[5]。草甘膦残留量的检测方法主要有气相色谱法、高效液相色谱法[6]、分光光度法[7]、酶联免疫法[8]、毛细管电泳法[9]、离子色谱法[10]和气相色谱-质谱法[11~12]等,笔者经反复实验,不断优化、改进,总结出一套水样中草甘膦和氨甲基膦酸气相色谱-质谱分析检测方法,且该方法操作简单,灵敏度高,重复性好,回收率高。
1 材料与方法
1.1 仪器与试剂
气相色谱-质谱联用仪:美国热电公司TRACE GC ULTRA-DSQ气相色谱-质谱联用仪,带电子轰击电离离子源(EI),Triplμs自动进样器,化学工作站;色谱柱:Agilent HP-INNOWAX石英毛细管柱(30 m×0.25 mm×0.25 μm);电子天平(感量 0.1 mg和 0.01 g各1台);德国2000型旋转蒸发仪,水浴锅;电热鼓风干燥箱;氮气吹干仪。
10 mL具塞刻度玻璃离心管、50 mL量筒、100 mL量筒、150 mL平底烧瓶、移液枪(10~50 μL和100~1 000 μL各 1只)。
三氯甲烷(色谱纯)、盐酸(分析纯)、甲醇(色谱纯)、柠檬醛(纯度≥95%)、三氟乙酸酐(TFAA)(分析纯)、七氟丁醇(HFB)(色谱纯)、乙酸乙酯(色谱纯)、氮气、蒸馏水。
农药标准品:99%草甘膦标准品、99%氨甲基膦酸标准品,均购于上海安谱实验科技股份有限公司。
1.2 气相色谱-质谱测定
1.2.1 气相色谱-质谱条件
柱箱采用升温程序:80℃保持1.5 min,以30℃/min升到260℃,保持2.0 min,进样口温度230℃,采用不分流进样,进样量为1 μL,载气为氦气,纯度99.999%,载气流量1.0 mL/min,传输线温度280℃,离子源温度250℃。采用选择离子监测(SIM),表1。

表1 保留时间、采集时间及离子监测
1.2.2 草甘膦和氨甲基膦酸的气相色谱-质谱定性
在上述色谱条件下,若样液中检出色谱峰的保留时间与标准溶液相一致,且被检出的样品质谱图与标准质谱图相似,所选择的检测离子均出现,其丰度比也相一致,相似度在20%左右时,则可确定待测物为草甘膦和氨甲基膦酸。
1.3 试剂溶液的配制
CAX洗脱液:分别量取160 mL去离子水、2.7 mL盐酸和40 mL甲醇,混匀。
0.2%柠檬醛乙酸乙酯溶液:100 mL乙酸乙酯中加入200 μL柠檬醛,混匀。
衍生试剂:三氟乙酸酐(TFAA)-七氟丁醇(HFB)(2+1,体积比),临用前配制。
草甘膦、氨甲基膦酸标准储备溶液:分别准确称取草甘膦标准品、氨甲基膦酸标准品10.0 mg于10mL容量瓶中,用去离子水定容到10 mL,配制成浓度为1.0 mg/mL的标准储备溶液,且转移至聚丙烯塑料瓶中,于0~4℃保存,有效期为1年。
草甘膦、氨甲基膦酸混合标准中间溶液:用CAX洗脱液将标准储备溶液分别稀释成10 μg/mL、50 μg/mL的混合中间溶液,存放于聚丙烯塑料瓶中,于0~4℃保存,备用。
草甘膦、氨甲基膦酸混合标准上机溶液:使用前用CAX洗脱液稀释混合标准中间溶液,配制成0.5 μg/mL的混合标准上机溶液。
1.4 测定方法和步骤
试样提取:取水样50 mL,放入200 mL具塞量筒,加三氯甲烷60.0 mL,振荡提取2 min,静置0.5 h。取上清液40~150 mL于平底烧瓶(浓缩瓶)中,45℃水浴中真空旋转浓缩近干,取出氮气吹干,用2 mLCAX洗脱液溶解样品,待衍生化。
衍生化:取三氟乙酸酐(TFAA)1.0 mL,七氟丁醇(HFB)0.5mL放入10 mL玻璃刻度离心管中,摇匀,放入-18℃冰箱中冷冻1 h,取出,用移液枪在衍生化试剂液面下缓慢加入样液50 μL,加盖后放入90℃电热鼓风干燥箱中衍生化反应1 h,其间每15 min轻摇1次,取出后冷却至室温,氮气吹干,并继续氮吹干0.5 h。加0.2%柠檬醛乙酸乙酯溶液溶解样品250 μL,充分溶解后,倒入250 μL进样瓶上机测定。
1.5 结果计算公式
按下式计算样品中草甘膦和氨甲基膦酸的残留含量。
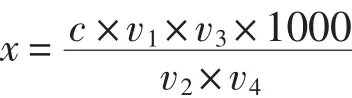
式中:x—样液中除草剂的含量,单位为μg/L;
c—上机样液浓度,单位为μg/mL;
v1—三氯甲烷提取体积,单位为mL;
v2—分取体积,单位为mL;
v3—最后定容体积,单位为mL;
v4—水样体积,单位为mL;
1000—单位换算系数,即:μg/mL换算为μg/L的系数。
本试验条件下,v1为 60 mL,v2为 40 mL,v3为 2 mL,v4为 50 mL。
2 结果与分析
2.1 色谱图
2.1.1 标准品色谱图
配制一定浓度的草甘膦、氨甲基膦酸的混合标准溶液,按1.2色谱-质谱条件测定,得到色谱图(图1)。
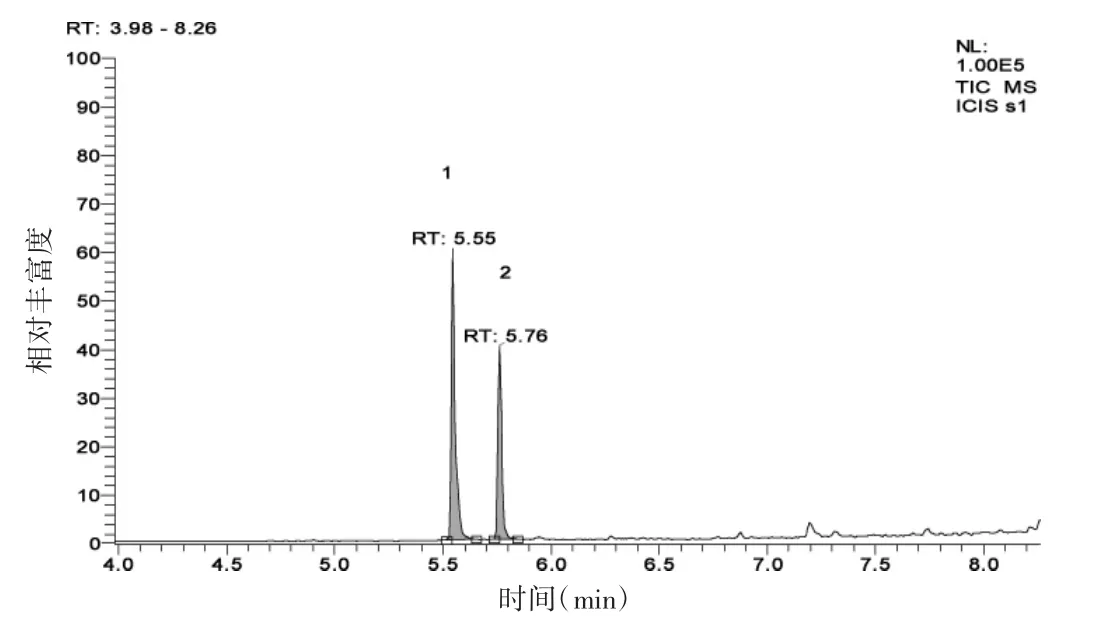
图1 氨甲基膦酸和草甘膦标准色谱图
由图1可知,在1.2优化的气相色谱-质谱条件下,氨甲基膦酸和草甘膦得到有效分离,且峰形好。
2.1.2 空白对照色谱图
除了不加入标准溶液,按1.4处理测定空白对照得到色谱图(图2)。
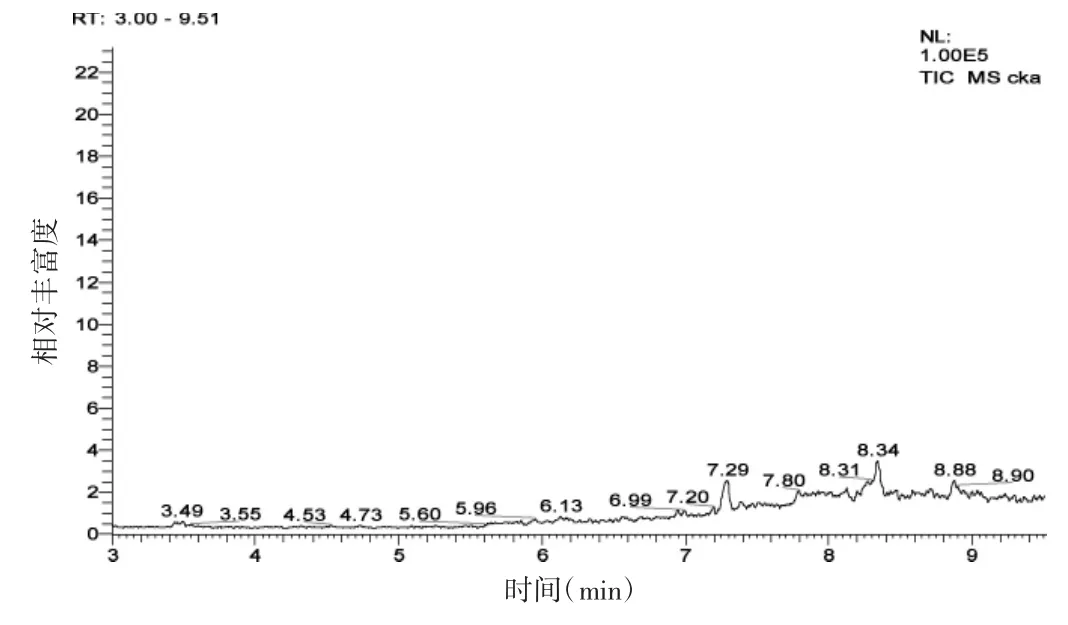
图2 水样空白
2.1.3 添加标样色谱图
在水样中添加草甘膦、氨甲基膦酸混合标准溶液,使水样中草甘膦、氨甲基膦酸浓度为25 μg/L,然后按1.4处理样品得到色谱图(图3)。
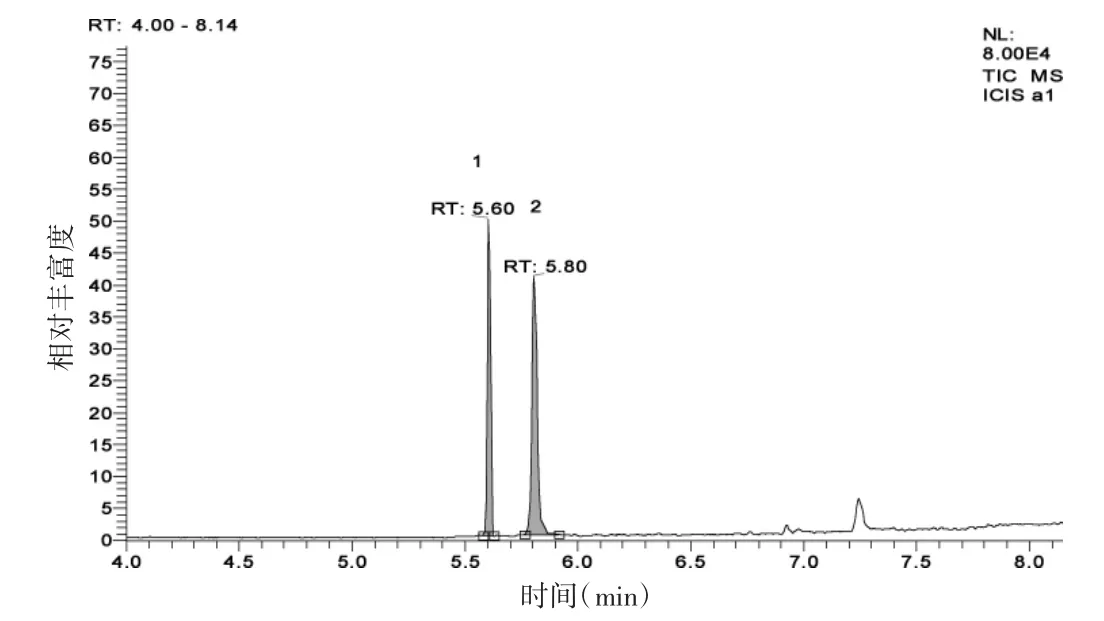
图3 水样中添加氨甲基膦酸和草甘膦标样色谱图
由图2、图3可看出,虽然在7.25 min处有一些杂峰,但不影响草甘膦和氨甲基膦酸的测定。
2.2 回收率、相对标准偏差、检出限测定
取60.0 mL水样4份,其中1份为对照,另3份加入浓度为10 μg/mL的草甘膦、氨甲基膦酸混合标准溶液 150 μL,即添加浓度为 25 μg/L,摇匀,放置 15 min,加入60.0 mL三氯甲烷振荡提取(以下同1.4),获得的色谱图按信噪比10倍条件计算方法检出限,结果见表2。
由表2可看出,氨甲基膦酸回收率在96.9%~120%,相对标准偏差为10.7%;草甘膦回收率在78.91%~93.3%,相对标准偏差为9.09%;在取样50mL,最后定容体积2 mL,进样量1 μL条件下,氨甲基膦酸方法检出限为0.6 μg/L,草甘膦方法检出限为0.5 μg/L,满足测定水样中草甘膦和氨甲基膦酸残留的需要。
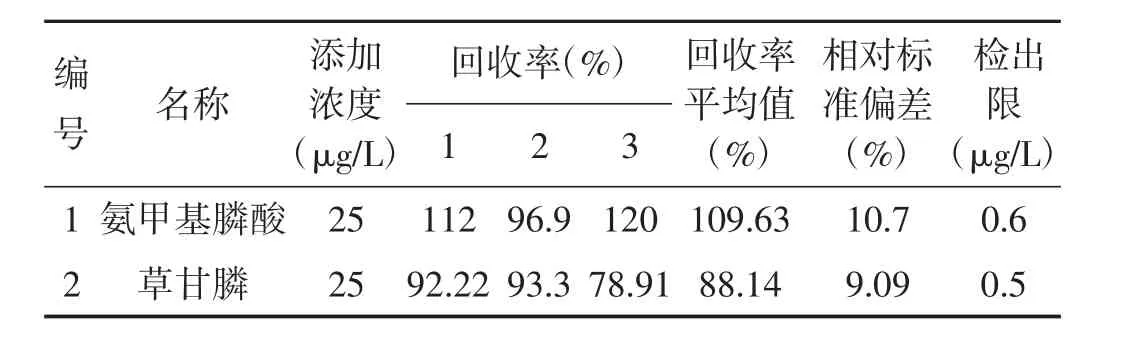
表2 回收率、相对标准偏差及检出限测定结果
3 结论
本试验采用三氯甲烷萃取,三氟乙酸酐(TFAA)和七氟丁醇(HFB)衍生化后,气相色谱-质谱联用仪检测,操作简单,对草甘膦、氨甲基膦酸定性准确,且准确度、精密度和灵敏度高,分析速度快,具有实用价值。
猜你喜欢
——以离子色谱法测定冬瓜中亚硝酸盐含量为例
现代食品(2022年15期)2022-09-08
波谱学杂志(2022年2期)2022-06-14
四川农业科技(2021年9期)2021-11-19
科技创新导报(2020年5期)2020-06-11
科教导刊(2017年26期)2017-11-07
今日农药(2017年9期)2017-09-30
科技与创新(2015年24期)2015-12-21
科技与创新(2015年17期)2015-09-11
科技与创新(2014年12期)2014-08-28
山东农药信息(2013年12期)2014-01-15
