
利用中压制备液相色谱从桑葚中快速制备矢车菊素-3-葡萄糖苷单体
2019-03-08 08:50冉国敬蒋鑫炜黎浩仪陈俊良李旭升孙建霞白卫滨
食品科学 2019年3期
冉国敬,蒋鑫炜,黎浩仪,陈俊良,李旭升,孙建霞,白卫滨,*
(1.暨南大学理工学院食品科学与工程系,广东省食品安全分子快速检测工程技术中心,广东 广州 510632;2.广东工业大学轻工化工学院,广东 广州 510090)
花色苷作为一类天然色素,其不仅安全、无毒、来源广泛,更为重要的是其具有的生物学价值。诸多研究已经表明花色苷对多重疾病有预防作用和健康促进功效,如抗氧化[1]、抗炎[2]、保护心血管[3]、改善生殖损伤[4]等。近年来随着对花色苷研究的逐步深入,对较高纯度花色苷尤其是单体花色苷的需求也逐步增长,然而当前其售价仍旧相当昂贵,市面上也鲜有成熟的单体花色苷制品,这主要是因为富含花色苷的原料比较单一以及提取、分离和纯化的成本太高。如何快速、大量、低成本地获得高纯度花色苷,尤其是单体花色苷,是目前花色苷开发应用面临的一个难题。
快速大量制备单体花色苷首先需要合适的原料。桑葚是为数不多的几种花色苷含量非常丰富的浆果之一。每100 g成熟桑葚可食用部分总花色苷质量可达185 mg[5],远高于众多其他果蔬,而其作为一种种植广泛且产量可观的鲜果,又极不耐贮藏和运输,这使得桑葚成为一种优良的花色苷提取潜在原料。而且很多研究表明桑葚中最主要的花色苷为矢车菊素-3-葡萄糖苷(cyanidin-3-glucoside,C3G),其含量可占到桑葚总花色苷的60%以上[6-8],这也更加证明以桑葚作为提取原料能够在很大程度上满足大量制备、生产单体花色苷的需求。
花色苷的分离纯化是获取单体花色苷的关键,现有研究中所使用的方法基本是凝胶色谱法[9-11]、高速逆流色谱法[12]、半制备高效液相色谱(high performance liquid chromatography,HPLC)法[13]等,但以上方法均存在单次处理量低、成本昂贵、耗时较长等不同缺点,很难在较短时间内快速得到大量的单体花色苷,具有一定的局限性。而中压制备液相色谱法是一种以大量制备为目的的快速色谱分离方法,其优点主要体现在单次处理量大、步骤简洁、分离高效、成本较低等方面[14-16],不仅在中药有效成分的快速分离和制备中应用较广,而且也已经成功地被应用于包括茶多酚中表没食子儿茶素-3-没食子酸酯单体[17]、柠檬苦素中诺米林酸-17-β-D-葡萄糖苷单体[18]、芦荟中芦荟苷A、芦荟苷B和异芦荟色苷D 3 种单体[19]以及栀子果实中京尼平苷、京尼平-龙胆双糖苷等6 种单体[20]在内的多种天然产物的分离纯化中,纯化得到的单体纯度均可达到95%以上。
本研究拟通过中压制备液相色谱法从桑葚中快速制得较大量C3G单体。在对桑葚中的总花色苷进行乙醇浸提、大孔树脂初步分离后采用中压制备液相色谱柱对粗分离产品进行进一步分离纯化,从而制备出C3G单体,并通过HPLC和质谱(mass spectrum,MS)进行纯度及结构确证。最终形成一种高效、简便、成本低廉的单体花色苷制备方法,为单体花色苷的分离纯化以及规模化生产提供良好的理论依据。
1 材料与方法
1.1 材料与试剂
桑葚果干来源于广东省农业科学院,室温保存;C3G标准品(纯度≥97%) 成都曼思特生物科技有限公司;Amberlite XAD-7HP大孔吸附树脂 美国Rohmand Haas公司;0.25~0.45 μm C18填料 日本富士硅化学公司;甲醇(分析纯) 上海泰坦科技股份有限公司;三氟乙酸(分析纯) 上海阿拉丁生化科技股份有限公司;其余试剂均为国产分析纯。
1.2 仪器与设备
LE104E电子分析天平 梅特勒-托利多仪器(上海)有限公司;RE-52AAA旋转蒸发仪 上海嘉鹏科技有限公司;DLSB低温冷却液循环泵 郑州长城科工贸有限公司;SHZ-D(III)循环水式真空泵 巩义市予华仪器有限责任公司;SB-5200DTS超声波清洗机宁波新艺超声设备有限公司;SCIENTZ-12N真空冷冻干燥机 宁波新芝生物科技股份有限公司;EZ PLUS 2000中压制备液相色谱仪(配备二元溶剂输送泵、紫外检测器及自动收集器) 利穗科技(苏州)有限公司;E2695分析型HPLC仪 美国Waters公司;8045 HPLC-MS/MS仪 日本岛津公司。
1.3 方法
1.3.1 桑葚花色苷粗提物的制备
称取一定量桑葚果干,去掉蒂部并洗净后按固液比1∶10加入体积分数65%酸化(体积分数0.1%三氟乙酸)乙醇溶液并打浆,浆液于42 ℃、200 W功率下超声40 min后静置12 h。采用布氏漏斗抽滤收集提取液,剩余浆体继续加入适量上述酸化乙醇溶液并超声浸提两次,合并3 次提取液于42 ℃减压旋转蒸发浓缩至无醇味后,加入1/3体积石油醚萃取,充分混合并避光静置4 h,收集下层水相,重复操作2~3 次直至萃取完全,下层水相于35 ℃减压旋转蒸发浓缩除尽石油醚后收集备用。将收集到的浓缩液缓慢加入到大孔吸附树脂柱(高100 cm、直径12 cm)中,单次上样体积为12 L,待其吸附后用2 倍柱体积(bed volume,BV)的0.1% HCl溶液以1 BV/h冲洗,再用体积分数80%乙醇溶液洗脱[21]。收集深色洗脱液,于42 ℃减压旋转蒸发浓缩至无醇味,再经冷冻干燥得到桑葚花色苷粗提品。整个提取、洗脱及浓缩过程均在避光条件下进行。
1.3.2 中压制备液相色谱柱填装及花色苷粗品的中压制备液相色谱分离纯化
中压制备液相色谱柱采用湿法填装:将约1.2 kg C18填料倒入甲醇中,充分混合摇匀后缓慢倒入中压制备色谱玻璃柱(高120 cm、宽6 cm)中,同时下端柱口连接真空泵进行减压抽气。待整个色谱柱填装好后连接保护柱并继续填装至填料高度到达保护柱1/2处,然后采用纯甲醇流动相进行柱体压实,压实完成后设置初始流动相条件和参数。取0.1 g花色苷粗品溶解到1 mL体积分数6%酸化(体积分数0.1%三氟乙酸)甲醇溶液中,混合均匀后上样进行预实验,根据预实验结果调整流动相梯度、流速等重要参数,逐步增加上样量至最大可处理量,经过多次调整后得到的最佳参数为花色苷粗品上样量4 g,流速36 mL/min。
取4 g桑葚花色苷粗品溶解到40 mL体积分数6%酸化(体积分数0.1%三氟乙酸)甲醇溶液中,混合均匀后上样。体积分数0.1%三氟乙酸溶液和甲醇分别为流动相A、B,检测波长500 nm,收集波长519 nm[22],监测波长280 nm和254 nm,收集阈值300 mAU,自动收集器采用多管收集方式进行洗脱液收集。按表1条件进行洗脱。

表1 中压制备液相色谱流动相洗脱梯度及流速Table1 Gradient elution program and fl ow rate for preparative MPLC
分别收集中压制备液相色谱分离得到的峰1~3洗脱液,减压旋转蒸发浓缩除尽甲醇后冷冻干燥,得到峰1~3的冻干粉末样品,备用。
1.3.3 HPLC分析
1.3.3.1 C3G标准品分析
取少许C3G标准品,用甲酸、水和甲醇(体积比5∶45∶50)溶液溶解,黑暗处平衡1 h,过0.22 μm有机滤膜后进行HPLC检测。
HPLC条件[23]如下:流动相A:5%(体积分数,下同)甲酸溶液;流动相B:5%甲酸-4 5%水-50%甲醇溶液;梯度洗脱条件:0~3 min,40%流动相B;3~27 min,40%~60%流动相B;27~32 min,60%~100%流动相B;32~40 min,100%~40%流动相B;色谱柱:Venusil ASB-C18(250 mmh4.6 mm,5 μm);柱温:30 ℃;流速:0~27 min,0.4 mL/min;27~32 min,0.4~0.8 mL/min;32~40 min,0.8 mL/min;检测波长:280 nm和520 nm;进样体积:10 μL;检测器:光电二极管阵列(photodiode array,PDA)检测器。
1.3.3.2 样品分析检测
分别取峰1~3冻干粉末样品,用适量流动相B溶解,过0.22 μm有机滤膜后按照1.3.3.1节条件进行HPLC检测。
1.3.4 MS分析鉴定
分别取峰1~3冻干粉末样品,适量超纯水溶解并稀释,过0.22 μm水系滤膜后进行MS分析鉴定,具体参数如下:电喷雾离子源;离子源温度:300 ℃;扫描范围:m/z 200~1 000,正离子模式;干燥气温度:300 ℃;干燥气流量:10.0 L/min。
对各样品进行Q3扫描后,选取各自丰度较高的分子离子峰进行MS/MS产物离子扫描,优化电压并分离特征离子峰,对其进行结构鉴定。
1.4 数据分析
采用利穗Chromatogragh workstation 1.41软件、Waters Empower软件和岛津LabSolutions LC-MS工作站软件进行数据分析和作图。
2 结果与分析
2.1 花色苷粗品的中压制备液相色谱分离纯化结果
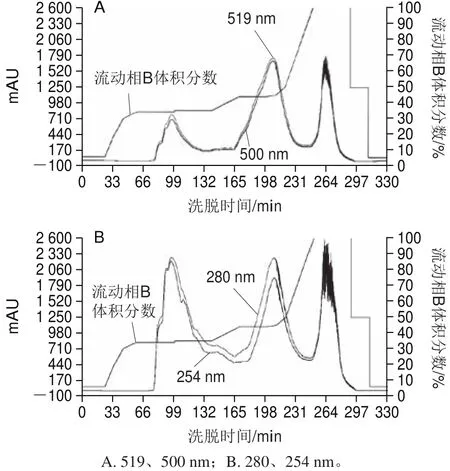
图1 中压制备液相色谱分离纯化花色苷粗品色谱图Fig.1 Separation chromatograms of crude anthocyanins by preparative MPLC
在最佳参数条件下中压制备液相色谱的分离纯化效果如图1所示,系统在519 nm波长处检测到响应阈值超过300 mAU后自动开始进行洗脱液收集,最终收集到3 个色谱分离峰所对应的3 种洗脱液,洗脱时间分别为79~115、170~226、252~277 min,3 个峰之间分离程度较好,峰形单一,可继续进行下一步纯度和成分的鉴定工作。
2.2 HPLC分析及MS鉴定结果
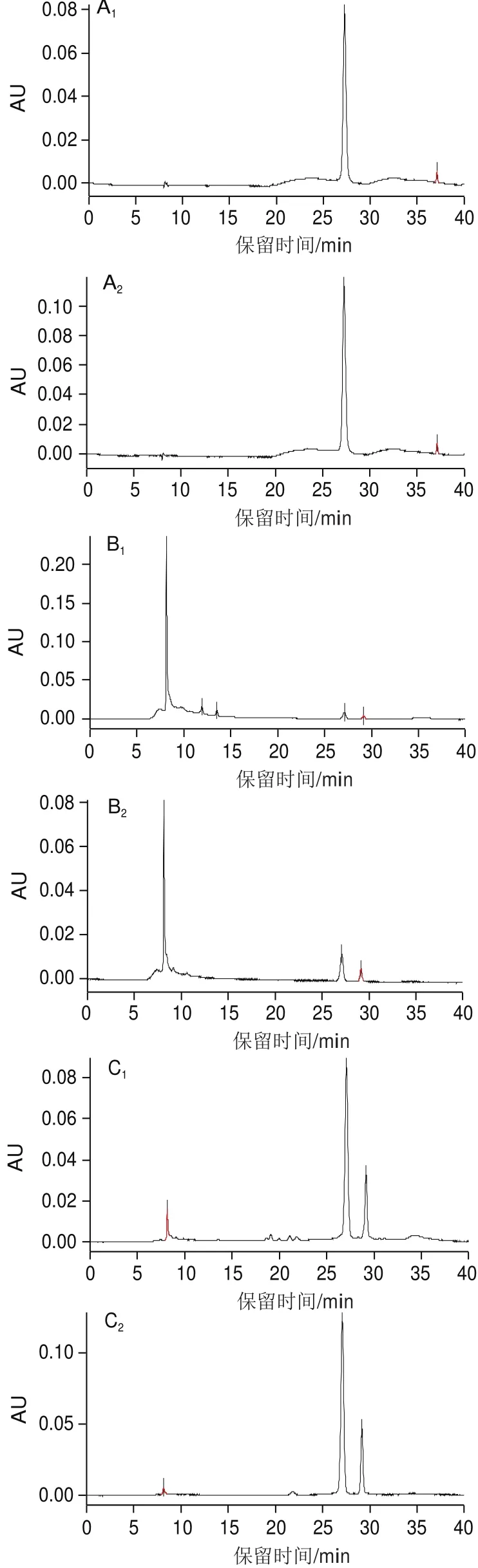

图2 C3G标准品(A)及中压制备液相色谱峰1(B)、峰2(C)、峰3(D)HPLC图谱Fig.2 HPLC chromatograms of C3G standard (A), eluate fraction 1 (B), 2 (C) and 3 (D)
由图2可知,峰1、2主要含有3 种物质,分别在8.1、27.1 min和29.1 min左右出峰;峰3相对比较杂乱,主要含有4 种物质,分别在8.1、27.1、29.1 min和35.9 min左右出峰。通过与C3G标准品的HPLC图对比发现,保留时间为27.1 min的物质与C3G标准品保留时间相对应,从而初步判定该物质为C3G。

图3 中压制备液相色谱峰1 MS扫描图Fig.3 Mass spectrum of eluate fraction 1 by preparative MPLC
通过MS/MS扫描各峰(图3~5)发现:峰1、3中组分相对杂乱,峰1主要分子离子为m/z 288.00、448.98和m/z 595.13;峰3主要分子离子为m/z 344.00、448.98和m/z 492.23;峰2中组分则较为单一,主要分子离子分别为m/z 448.98和m/z 595.03。对m/z 448.98的分子离子在15 V电压下碰撞后进行产物离子扫描,由图4B可知,碎片离子m/z 287为矢车菊素,由母离子(m/z 449)通过失去一个葡萄糖苷(m/z 162)中性碎片而形成[24],由此判断m/z 448.98的分子离子所代表的物质为C3G。对m/z 595.03的分子离子在18 V电压下碰撞后进行产物离子扫描,由图4C可知,碎片离子m/z 287为矢车菊素,m/z 449为C3G,分别由母离子(m/z 595)通过失去一个芸香糖苷(m/z 308)中性碎片和一个鼠李糖苷(m/z 146)中性碎片而形成[25],由此判断m/z 595.03的分子离子所代表的物质为矢车菊素-3-芸香糖苷(cyanidin-3-rutinoside,C3R)。对m/z 492.23的分子离子在19 V电压下碰撞后进行产物离子扫描,由图5B可知,碎片离子m/z 330为锦葵素,由母离子(m/z 492)失去一个葡萄糖苷(m/z 162)中性碎片而形成[13],从而判断m/z 492.23的分子离子所代表的物质为锦葵素-3-葡萄糖苷(malvidin-3-glucoside,M3G)。结合HPLC和MS/MS分析结果最终判断峰1、2中主要花色苷成分为C3G和C3R,峰3中主要花色苷成分为C3G和M3G。以峰2作为目的峰,通过峰面积积分法计算得到在280 nm检测波长处峰2中C3G纯度为73.56%。

图4 中压制备液相色谱峰2 MS/MS扫描图Fig.4 Tandem mass spectra of eluate fraction 2 by preparative MPLC

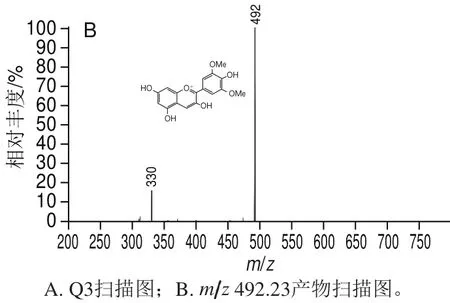
图5 中压制备液相色谱峰3 MS/MS扫描图Fig.5 Tandem mass spectra of eluate fraction 3 by preparative MPLC
由于中压制备液相色谱系统收集洗脱液时采用的是多管收集方式,每管可单独收集50 mL洗脱液,为进一步制备C3G单体,对于峰2中未完全分离的两种花色苷尝试采用切割法进行流分收集[26]。对峰2整个收集区域进行如图6所示的切割分段取样后用HPLC检测,结果如图7所示。随着收集时间的延长,C3G纯度和比例逐步发生变化,在280 nm波长处4 个区域中的C3G纯度分别为98.72%、69.07%、66.62%和13.43%,其中1号区域C3G纯度达到98%以上,经Q3扫描发现1号区域组分单一,同时MS/MS也确证分子离子m/z 448.98即为C3G(图8)。最终选取1号区域作为切割收集区,单次即可收集到650 mL(13h50 mL)溶液,经旋转蒸发浓缩和真空冷冻干燥后成功制备出C3G单体。

图6 中压制备液相色谱各峰对应收集区域及取样区段Fig.6 Fractional collection of peaks separated by preparative MPLC

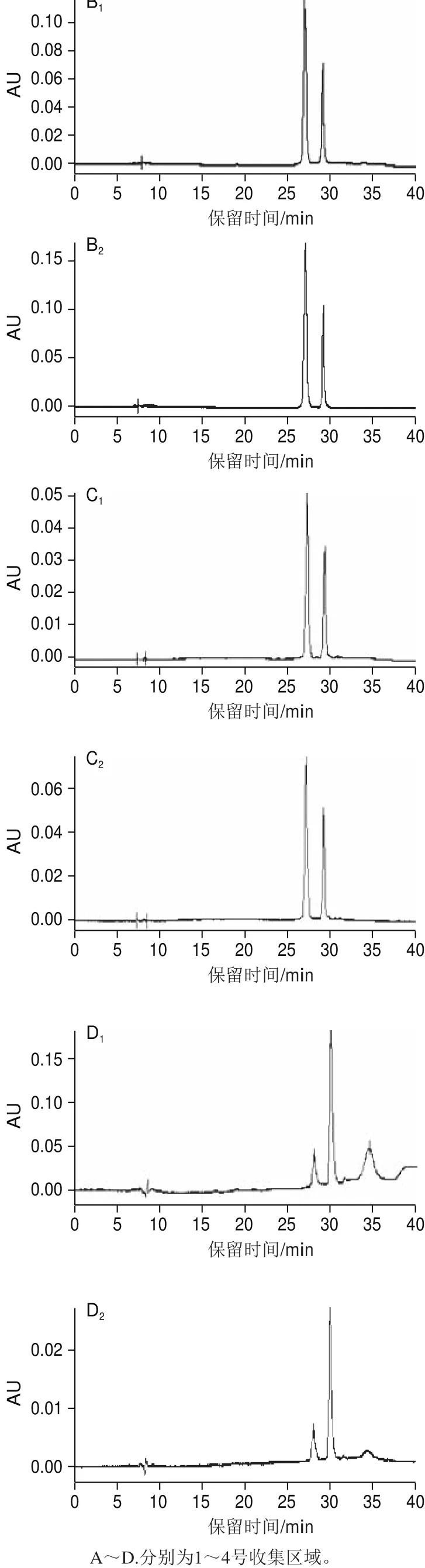
图7 峰2不同收集区域HPLC图Fig.7 HPLC chromatograms of fractions separated from peak 2

图8 1号收集区域MS/MS扫描图Fig.8 Tandem mass spectra of fraction 1
3 讨 论
本研究以体积分数0.1%三氟乙酸酸化的体积分数65%乙醇溶液作为提取液,采用超声波辅助提取法提高桑葚花色苷的提取效率。由于桑葚提取液中包含部分脂溶性杂质,使用石油醚萃取除去提取液中的色素、挥发油、脂肪酸等脂溶性成分。
实验前根据花色苷的极性选取合适的C18填料,然后对中压制备液相色谱柱进行填装,一次填装即可长时间使用,无需每次重复装柱。在对桑葚花色苷粗品进行分离纯化时,通过多次的参数条件调整,发现上样量为4 g时可达到最佳分离纯化效果,如果再增加上样量则无法达到较好的分离效果。最佳条件下共得到3 个色谱分离峰,其中以峰2作为目的峰,通过HPLC和MS检测鉴定出峰2中主要物质为C3G和C3R,纯度分别为73.56%和20.96%,峰2整体C3G纯度还达不到单体标准[27]。通过查阅文献[28]发现,花色苷的HPLC行为常表现为二糖苷出峰在一糖苷之后,而C3G和C3R这两种单体花色苷的区别正好在于此,本研究中的HPLC出峰结果也与之相符。实验中也曾通过继续优化条件来尝试将二者完全分离,但未能到达预期效果。而由于中压制备液相色谱收集洗脱液时使用多管收集方式,非常适合采用切割法对峰2进行分段流分收集,最终经检测鉴定后切割收集区的C3G纯度达到98.72%,成功制备出C3G单体。此外,除了得到目标单体,峰1、3也可作为副产物收集后用于进一步纯化得到其他单体物质。
相较于其他纯化制备技术,中压制备液相色谱不仅在处理量及处理效率上要优于高速逆流色谱、半制备HPLC、柱层析等方法,而且也同样能制得高纯度的单体花色苷。高速逆流色谱纯化0.1 g粗品需250 min,可得纯度为98.1%的C3G单体[29];半制备HPLC虽然能在20 min内纯化得到2 种纯度均高于98%的单体花色苷,但是其单次最佳处理量只能达到0.035 g[30];硅胶柱层析法平均纯化0.275 g粗品需要300 min,所得两种单体花色苷纯度均只能达到82%以上[31];而本研究中所使用的中压制备液相色谱法能在320 min内纯化4 g粗品,所制得的C3G单体纯度可达到98.72%。同时,中压制备液相色谱流动相只需使用分析纯试剂,且只用到甲醇一种有机试剂,后期通过旋转蒸发即可除去,整个方法绿色、经济、简洁,能够在1 d内连续处理8~12 g粗品,每天可截取收集到1 300~1 950 mL C3G单体溶液,经浓缩和干燥即得到C3G单体。在C3G单体的大量制备以及更大规模的工厂化生产上都具有很大的开发空间。
4 结 论
以桑葚作为原料,采用超声波辅助酸化乙醇浸提法提取花色苷,通过石油醚萃取及大孔树脂吸附除杂后使用中压制备液相色谱分离纯化花色苷粗品,并利用HPLC和MS技术对各峰物质进行鉴定。结果显示:桑葚中主要花色苷成分为C3G、C3R和M3G,目的峰(峰2)整体C3G纯度为73.56%,对峰2进一步采用切割方式收集后C3G纯度上升到98.72%。单次上样量达4 g,可切割收集到650 mL洗脱液,经浓缩冻干即得C3G单体,本方法处理量大、简便易行、绿色高效,在花色苷单体的大量制备及产业化生产上具有应用前景。
猜你喜欢
陕西科技大学学报(2022年6期)2022-12-01
食品工业科技(2021年17期)2021-09-14
落叶果树(2021年6期)2021-02-12
课堂内外(高中版)(2021年7期)2021-01-17
阅读与作文(小学低年级版)(2020年11期)2020-12-21
启蒙(3-7岁)(2019年4期)2019-06-27
中国生殖健康(2019年9期)2019-01-07
天然产物研究与开发(2018年5期)2018-06-13
红蜻蜓·低年级(2017年8期)2017-10-30
山西青年(2017年16期)2017-09-03
