
Al (1 1 1) /Al3Li (1 1 1)的界面性质
2019-04-29 03:08孔德斌潘荣凯尹登峰
原子与分子物理学报 2019年2期
孔德斌, 潘荣凯, 尹登峰, ,
(1. 烟台南山学院工学院,烟台 265713; 2. 中南大学材料科学与工程学院,长沙 410083; 3. 中南大学,教育部有色金属材料重点实验室,长沙 410083)
1 引 言
L12-Al3Li金属间化合物作为第三代Al-Li合金的重要析出相,能有效改善铝合金的强度和高温抗蠕变性能,其时效行为和基本热力学性能近几十年来一直铝合金重点研究领域之一[1-5].研究表明,Al-Li合金的高弹性模量与Al/Al3Li界面特征密切相关[6].因此,研究Al-Li合金析出相(δ′Al3Li等)与基体Al之间的界面性质具有重要的意义.事实上,Baumann, Mao等已经分别从理论和实验上给出了Al/Al3Li的界面能[7, 8].Gao 等不仅从原子成键的角度,计算了Al3Li相的价电子结构[9],而且运用固体经验电子理论的电子密度分析法,对比计算了δAl3Li和δ′Al3Li相与基体之间的界面电子性质的差异[10],认为Al/δ′Al3Li的界面电子密度在较低应力下保持连续,从而说明δ′Al3Li相与基体结合较好,起到了增强界面的效果.我们认为,研究Al/Al3Li的界面性质,首先应该确定Al和Al3Li形成界面时界面取向, 同一种界面取向,还应该考虑析出相与基体的原子配位关系.同时,基体与Al3Li相的界面附近不同的区域,强度显然也是不一样的.界面能只是映了形成界面的难易程度,电子密度分析法也只是间接说明界面区域强度的方法.鉴于此,本文运用密度泛函理论,以Al (111)/ δ′Al3Li (111) 界面取向为例,计算给出了不同的原子匹配关系形成的界面构型.同时用分离功的方法探讨最稳定构型的界面附近层与层之间的强度,希望从原子层面反映δ′Al3Li强化合金性能的内在原因.
2 计算方法
所有计算是在基于密度泛函理论的VASP[14]程序中进行的.选择投影缀加平面波赝势(PAW)[15]来描述离子—电子间的相互作用,采用广义梯度近拟(GGA)中的PBE[16]方法处理电子间的交互关联作用.在计算Al, Li, Al3Li单胞模型以及Al3Li三个低指数面表面时,简约布里渊区的 K点网格采用Monkhorst-Pack方法(16×16×16)来划分, 而Al (111) /Al3Li (111) 界面性质的计算则采用8×8×1的K点.所有单胞及表面,界面模型的波函数动能截断能取320eV.驰豫能量和原子力收敛判居分别为1×10-4和0.02eV/Å.
3 结果与讨论
首先对Al, Li, 及L12-Al3Li 单胞进行了包括体积和原子坐标在内的几何优化. 通过 Birch-Murnaghan状态方程拟合方法得到Al, Li, 及L12-Al3Li 晶格常数 、体模量、基态能(见表1).计算结果与实验值接近,表明本文采用的计算参数是合理的.
表1Al, Li, 和L12-Al3Li的晶格常数 、体模量、基态能的计算值和实验值
Table1The calculated results and experimental data of lattice constant, bulk modulus, and ground state energy for Al, Li, and L12-Al3Li.

Elementa/ÅE0/evB/GPaAl4.041, 4.05 [17]-3.74577.7, 76 [17]Li3.51, 3.44 [17]-1.89913.998, 11 [17]Al3Li4.028, 4.010 [18]-13.5263.91, 66 [18]
本文确定Al (111) /Al3Li (111)的界面模型时,不仅要确定构成界面的表面的原子匹配关系,还要确定构成界面的原子层数,以及真空层的厚度.
不同的结构由于晶格常数的差异,形成界面时,在界面处会产生失配位错.因此,在构建界面模型时,应该将界面取得足够大,将界面处的变形均匀地引入其中.但是界面太大,会增加计算量.鉴于此,我们在界面附近的晶格施加了尽量小的应变,使之发生一定的合理变形,以此形成理想的共格界面模型.本文界面处两侧Al和δ′(Al3Li)体模量不同,我们据此设计了不同的变形量.Al(111)面和Al3Li(111)面的应变量分别为压缩0.11%和拉伸0.22%,应变在界面的方向上是均匀的.
真空层的厚度对界面计算也有影响.厚度太小,原子在Z方向可能驰豫不充分,影响计算结果.厚度太大,会使计算量太大.本文选择12Å的真空层,既可以保持结果准确性,也可以避免计算资源的浪费.
在FCC结构中,Al(111)和Al3Li(111)面都是按照ABC顺序堆垛的,因此很容易判断Al(111)面和Al3Li(111)面有三种配位关系,分别命名为Inter-A, B ,C,如图1所示.
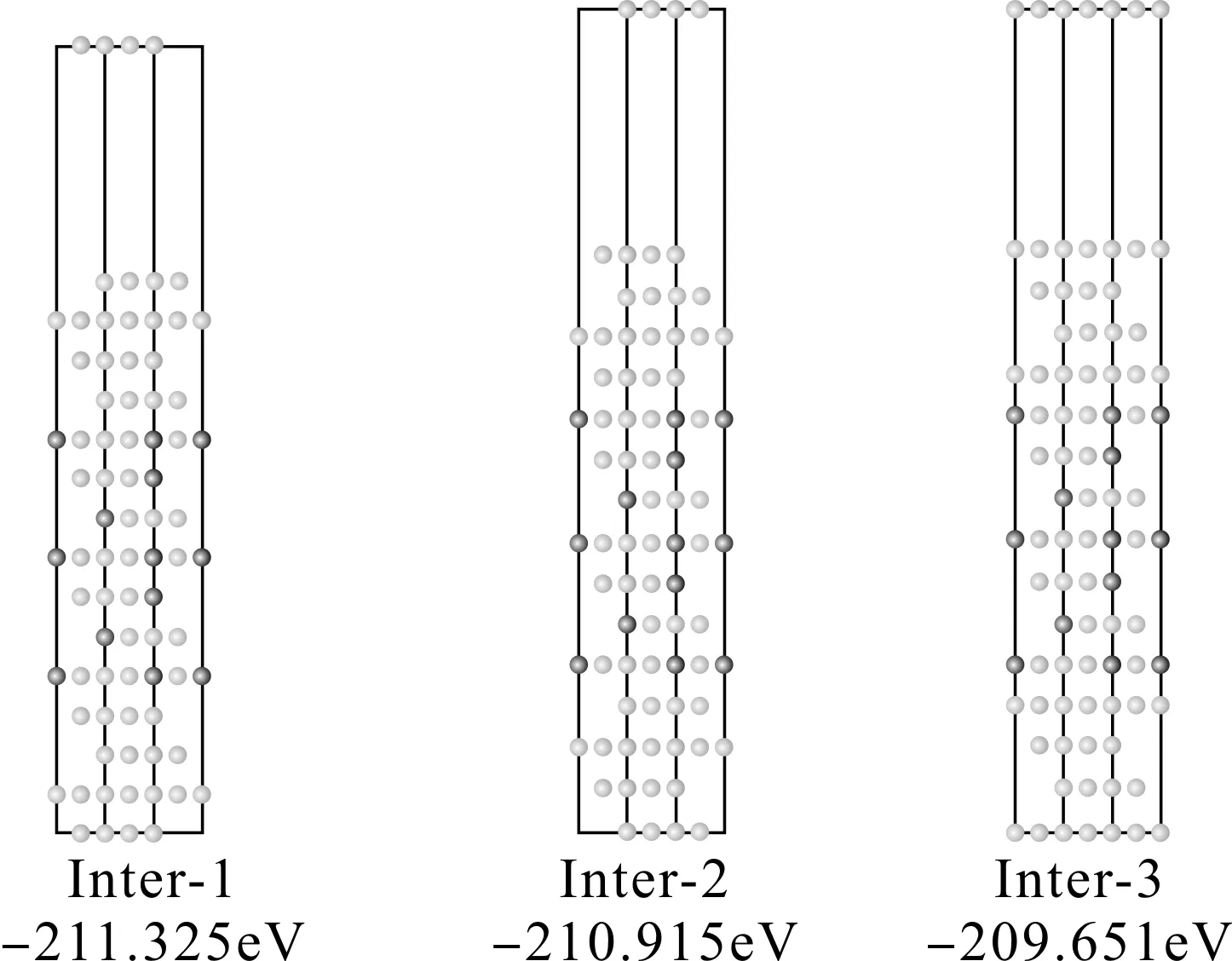
图1 Al (1 1 1) /Al3Li (1 1 1) 界面三种配位关系示意图 (白色为Al原子,黑色为Li原子)Fig. 1 Sketch maps of three coordination relations for Al (1 1 1) /Al3Li (1 1 1) interface (blue and green colors represent Al and Li atoms, respectively).
本文对上述三种三明治形的共格Al/Al3Li/Al界面模型分别进行了驰豫计算.整个界面超胞模型包含上下各4层Al原子,中间7层Al3Li和12Å的真空层.采取中间层固定,上下层同时驰豫的方法.计算结果表明,以上三种配位关系的界面模型中,Inter-1配位关系的界面超胞总能最低 (如图2所示),而且驰豫前后界面结构基本保持不变.自由能越低越稳定,所以可以确定Al/Al3Li/Al界面最稳定的配位关系为Inter-1.这是可以理解的.因为Inter-1界面处原子排布仍然采用的是ABC顺序堆垛的三明治结构,与基体Al完全一致,对于FCC结构,这种原子排布更有利于结构稳定,所以能量最低.

图2 Al和Al (1 1 1) /Al3Li (1 1 1) 界面的差分电荷密度(浅色表示电荷增加,深色表示电荷减少)Fig. 2 The deformation charge density Al and Al (1 1 1) /Al3Li (1 1 1) interfaces (yellow and blue colors represent the increase and decrease of the charge, respectively).
为了进一步说明成键时界面处的特性与基体Al的差别,我们在图2给出了Al (111) 面以及Al (111) /Al3Li (111) 三种界面的差分电荷密度图.可以看出,Al(111)面的层与层之间电荷均匀分布,表现出了金属键的特性.而对于Al (111) /Al3Li (111)三个界面,不仅界面处成键特性发生了改变,界面附近区域电荷分布也与基体Al存在差别.不过,相比与Inter-2和Inter-3,Inter-1不论是界面处,还是界面附近,电荷分布与基体Al的分布最为接近,因此其稳定性应该是最好的,这与上文能量计算的结果是一致的.
本文采用界面粘附功Wads和界面分离功Wsep来定量研究界面的结合强度.界面粘附功是两个自由表面结合成一个界面所放出的能量,结合前后原子都充分驰豫到稳定状态.界面分离功是界面瞬间断裂成两部分所需提供的能量,断裂后,原子来不及发生驰豫.前者一般可以反映生成界面两相的润湿性,后者可以用来评价界面的断裂强度.一般的材料,断裂往往发生在分离功最低的地方.但是需要注意的是,分离功衡量的是整个界面区域的强度,不仅仅只反映界面处的结合强度.粘附功和分离功的计算公式定义如下:

图3 Al(1 1 1)/Al3Li(1 1 1)界面粘附功Fig. 3 Adsorption work of Al (1 1 1) /Al3Li (1 1 1) interface.

图4 Al(1 1 1)/Al3Li(1 1 1)界面分离功. (1) 界面各层分离功 (2) 界面在最薄弱层处发生断裂的结果Fig. 4 Separation work of Al (1 1 1) /Al3Li (1 1 1) interface.(1) Separation work for each layer. (2)The results of fracture for the weakest position of the Al (1 1 1) /Al3Li (1 1 1) interface.
本文计算的粘附功为1.588J/m2,但这只是反映了界面结合时的难易程度.为了说明整个界面区域的强度,我们计算了界面处5个可能的断裂面的分离功,结果如图4所示.图4(1)给出了界面可能的断裂面的位置.需要说明的是,每一个断裂面的位置都是上下对称的,我们给出的仅是界面的上半部分.图4(2)展示的界面在最薄弱层断裂的结果.可以看出,最薄弱层发生在Al3Li内部,计算结果表明,该处的分离功值是最小的,为1.53eV.而基体Al内部的分离功随着离界面距离增大,其值逐渐增大.Al3Li内部的各层结合强度表现也出了相似的变化规律.
4 结 论
Al (111) / Al3Li (111) 的界面具有三种原子配位关系结构,界面处仍保持与基体Al相同的三明治堆垛结构的模型,驰豫后能量最低.相比于其它两种结构,其界面处及界面附近电荷分布与基体Al相比,变化不大.计算表明,该结构强度最弱处位于Al3Li内部.基体Al及Al3Li内部的分离功随距离界面的距离的增加而逐渐增大.
猜你喜欢
石材(2022年3期)2022-06-01
原道(2022年2期)2022-02-17
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
理化检验-化学分册(2020年5期)2020-06-15
当代陕西(2019年6期)2019-04-17
橡胶工业(2015年8期)2015-07-29
外语学刊(2014年3期)2014-12-03
