
阻断辅因子NADPH合成对谷氨酸棒杆菌生长及产物合成的影响
2019-06-06 06:51杨汉昆徐建中张伟国
食品与发酵工业 2019年10期
杨汉昆,徐建中*,张伟国*
1(江南大学 生物工程学院,江苏 无锡,214122) 2(工业生物技术教育部重点实验室(江南大学),江苏 无锡,214122)
还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate, NADPH)又称为还原型辅酶Ⅱ,在很多生物体内的化学反应中起递氢体的作用,具有重要的生物学意义。NADPH在细胞内分布广泛,通过参与800多个氧化还原反应来调节细胞内氧化还原水平并影响着众多基因表达、细胞功能、代谢途径和物质跨膜运输[1],参与多种合成代谢反应,如氨基酸、脂类及核苷酸等细胞组成物质的合成均需要NADPH提供还原力,对细胞正常生长和代谢有重要影响[2],并且是微生物代谢网络中含量最丰富的氧化还原辅酶之一。胞内NADPH的生成和消耗同胞内很多重要的代谢途径相关联,维持细胞内辅酶的平衡对于细胞生长、代谢以及产物的合成都非常关键。烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide, NAD)是另一种极为重要的核苷酸类辅酶,是代谢网络中的关键辅因子之一,在胞内参与超过300个氧化还原反应[3]。如在糖酵解、糖异生、三羧酸循环以及呼吸链等代谢中发挥着不可替代的作用[4-6]。NADH/NAD+这对辅因子在葡萄糖分解代谢中起着核心作用,以NAD+作为辅因子将葡萄糖氧化,并且NAD+同时转化成等量的NADH还原形式。
NADPH对谷氨酸棒杆菌(C.glutamicum)合成L-赖氨酸非常重要,根据C.glutamicumL-赖氨酸合成代谢网络可知有4个反应涉及NADPH的消耗,即合成1 molL-赖氨酸需要消耗4 mol NADPH[7]。研究结果表明在谷氨酸棒杆菌中NADPH的供应与消耗是不平衡的。在谷氨酸棒杆菌中分别由葡萄糖-6-磷酸脱氢酶、6-磷酸葡萄糖酸脱氢酶、苹果酸酶和异柠檬酸脱氢酶以NADP+为辅因子,参与NADPH合成[8-9]。谷氨酸棒杆菌中的NADPH不仅要满足L-赖氨酸合成需求而且还要用于菌体生长。因为,增加1g菌体需要消耗16.4 mmol NADPH[10]。目前的研究主要集中于通过辅因子工程提高NADPH的供应可以强化L-赖氨酸合成,而对阻断胞内NADPH合成途径对菌体生长及产物合成的影响研究匮乏。胞内NADPH水平可以改变胞内微环境,CHEN等[11]指出,改变胞内NADPH水平会扰动胞内的氧化还原水平和ATP含量;同时也会影响目标代谢产物产量,XU等[12]通过在谷氨酸棒杆菌中异源表达大肠杆菌膜结合转氢酶PntAB来改变胞内NADPH水平,能够影响谷氨酸棒杆菌合成L-赖氨酸的能力。氧化还原辅因子代谢工程成为优化生物转化的重要代谢工程策略[13]。在谷氨酸棒杆菌中,研究表明L-赖氨酸的合成与辅因子NADPH水平密切相关[1]。
本研究以实验室保存的L-赖氨酸生产菌株C.glutamicumLYS为出发菌株,敲除参与胞内NADPH合成的葡萄糖-6-磷酸脱氢酶、苹果酸酶编码基因,并将自身NADP+-依赖型异柠檬酸脱氢酶基因(icdCg)替换成变形链球菌(Streptococcusmutans)NAD+-依赖型异柠檬酸脱氢酶基因(icdSm)。对出发菌C.glutamicumLYS和重组菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm进行摇瓶实验,分析阻断谷氨酸棒杆菌中NADPH合成途径对胞内NAD+、NADH、ATP、ADP和AMP、细胞生长、葡萄糖消耗速率、L-赖氨酸、有机酸以及其他副产物氨基酸合成的影响。为进一步研究NADPH调控L-赖氨酸产生菌胞内微环境的生理机制,调控和优化胞内NADPH与中心碳代谢的特异性,为改进微生物L-赖氨酸生产性能提供了研究基础。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株和质粒
C.glutamicumLYS、E.coliJM109,穿梭表达质粒pDXW-8和敲除质粒pK18mobsacB均为本研究室保藏,其他质粒均为实验中构建。
1.1.2 主要试剂和仪器
限制性内切酶BamH Ⅰ、XbaⅠ、Hind Ⅲ、SalⅠ、EcoR Ⅰ,T4DNA连接酶、Taq DNA聚合酶、DNA Marker:购于宝生物工程有限公司;PCR相关试剂、基因组DNA提取试剂盒、DNA凝胶回收试剂盒、PCR产物纯化试剂盒、质粒小量提取试剂盒:购于上海生工生物工程有限公司;辅酶Ⅱ NADP(H)和NAD(H)含量检测试剂盒:购自北京索莱宝科技有限公司;其他试剂均为进口或国产分析纯试剂。
UV 2100可见紫外分光光度计,尤尼柯(上海)仪器有限公司;JY 1600C凝胶水平电泳仪,北京六一仪器厂;SBA40-E生物传感仪,山东省科学院生物研究所;G1-14高速离心机,Sigma公司;Gel DOC GR+凝胶成像系统,美国Bio-Rad公司;LC 1100高效液相色谱系统,安捷伦科技有限公司;Gene Pulser Xcell电穿孔仪,美国Bio-Rad公司。
1.1.3 培养基
LB培养基(g/L):蛋白胨 10,酵母粉 5,NaCl 10,pH 7.0。
LBG培养基(g/L):蛋白胨 10,酵母粉 5,NaCl 10,葡萄糖 5,pH 7.0。
Epo培养基(g/L):葡萄糖 5, 蛋白胨 10, 酵母膏 5, NaCl 10, 吐温-80 1, pH 7.0,121 ℃灭菌20 min, 灭菌后加入无菌的异烟肼4 g和甘氨酸30 g,用于制备C.glutamicum感受态细胞。
LBHIS培养基(g/L):蛋白胨 5,酵母膏 2.5,NaCl 10,脑心浸出液 18.5,山梨醇 91,pH 7.0,121 ℃灭菌20 min,用于C.glutamicum转化子的恢复和生长。
种子培养基(g/L):豆粕水解1.25,(NH4)2SO42,蔗糖 25,KH2PO41, MgSO4·7H2O 0.4,乙酸铵 6,FeSO40.01,MnSO40.01,烟酰胺 0.005,味精 0.1,硫胺素0.000 2,生物素0.000 5,121 ℃灭菌20 min,500 mL三角瓶装液量50 mL,30 ℃、100 r/min培养16 h。
发酵培养基(g/L):葡萄糖 40,(NH4)2SO436,MgSO4·7H2O 1.5,FeSO40.02,MnSO40.02,KH2PO41,K2HPO41,玉米浆 35,甜菜糖蜜 12,烟酰胺 0.008,甜菜碱0.05,硫胺素0.000 45,生物素0.000 85,CaCO340,pH 7.0,115 ℃灭菌10 min。5 mL种子液转接到装有50 mL发酵培养基的500 mL的摇瓶中,30 ℃、 100 r/min发酵40 h。
1.1.4 PCR引物序列
基于NCBI数据库中C.glutamicumATCC 13032基因组序列设计用于各基因敲除框的扩增和验证引物,基于StreptococcusmutansJH1005基因组设计用于扩增置换基因的引物。如表1所示。
1.2 实验方法
1.2.1 胞内NADPH合成相关基因的敲除载体pK18-mobsacB-Δzwf、pK18mobsacB-ΔmalE的构建
根据NCBI中C.glutamicumATCC 13032基因序列设计zwf基因上下游以及中间引物,引物序列如表1。提取C.glutamicumATCC 13032基因组为模板,分别以zwf-L-F/zwf-L-R和zwf-R-F/zwf-R-R为引物PCR,获得分别在3’端和5’端有相同限制性内切酶酶切位点的PCR产物zwf-L、zwf-R。将胶回收后的基因片段zwf-L和重组自杀型质粒载体pK18mobsacB通过HindIII和SalI双酶切,纯化后将zwf-L和线性化质粒pK18mobsacB22 ℃过夜酶连,转化E.coliJM109,经抗性平板筛选,挑取转化子菌落PCR,选取较亮条带对应的单菌落液体培养后提取质粒酶切验证,构建质粒pK18mobsacB-Δzwf-L。用同样的方法将基因片段zwf-R连接至质粒pK18mobsacB-Δzwf-L上构建重组自杀型质粒pK18mobsacB-Δzwf。通过相同的方法构建质粒pK18mobsacB-ΔmalE。
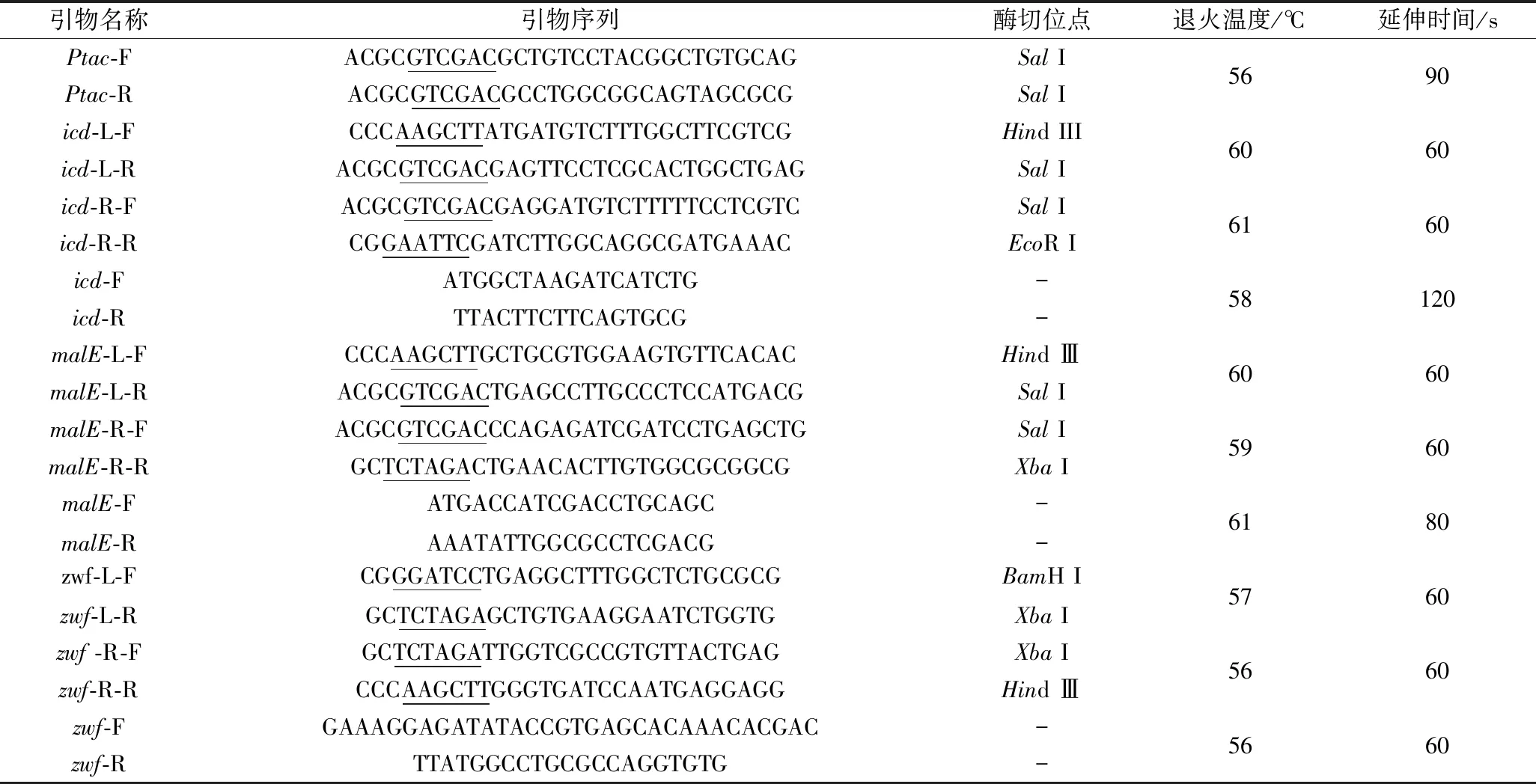
表1 PCR扩增所需引物序列Table 1 Primer sequences required for PCR amplification
注:下划线为酶切位点。
1.2.2 胞内NADPH合成相关基因的置换载体pK18mobsacB-ΔicdCg::icdSm的构建
通过1.2.1中的方法构建基因敲除质粒pK18mobsacB-ΔicdCg。
根据NCBI中StreptococcusmutansJH1005全基因组核酸序列中的icd基因序列,在其基因上下游分别加入限制性内切酶EcoR I和XhoI酶切位点序列并在上游加入谷氨酸棒杆菌SD识别序列GAAAGGAGATATACC,并将组合好的序列提交给通用生物系统(安徽)有限公司进行合成,获得含有目的基因的重组质粒pUC57-icdSm。
采用限制性内切酶EcoR I和XhoI酶切重组质粒pUC57-icdSm。随后采用胶回收试剂盒回收icdSm片段。将icdSm片段与经相同限制性内切酶酶切后的C.glutamicum-E.coli穿梭表达质粒pDXW-8相连构建重组质粒pDXW-8-icdSm。
根据表达质粒pDXW-8基因序列设计包含启动子Ptac和终止子rrnBT1T2基因上下游引物,引物序列如表1。以pDXW-8-icdSm模板,以Ptac-F/Ptac-R为引物进行PCR,获得Ptac-icdSm-rrnBT1T2表达框。随后将纯化后的PCR产物Ptac-icdSm-rrnBT1T2与pK18mobsacB-ΔicdCg用SalI单酶切,酶切产物纯化后酶连,获得在icdCg左右同源臂间插入icdSm基因,构建基因置换质粒K18mobsacB-ΔicdCg::icdSm。
1.2.3 重组菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm的构建
1.2.4 胞内辅酶Ⅱ NADP(H)的测定[16]
收集400~500万细菌,加入0.9 mL酸性(碱性)提取液,超声破碎1 min(200W,超声2 s,停1 s),盖紧后煮沸5 min,冰浴中冷却后,10 000×g4 ℃离心10 min,取上清液200 μL至另一新的离心管中,加入等体积的碱性(酸性)提取液使之中和,10 000×g、4 ℃离心10 min,取上清。随后,以试剂盒NADP/NADPH Quantification Colorimeteric Kit特异性检测NADP+和NADPH,并计算NADPH /NADP+。
1.2.5 胞内辅酶Ⅰ NAD(H)的测定[16]
收集500万细菌,加入0.5 mL酸性(碱性)提取液,超声破碎1 min(200W,超声2 s,停1 s), 盖紧后煮沸5 min,冰浴中冷却后,10 000×g4 ℃离心10 min,取上清液200 μL至另一新的离心管中,加入等体积的碱性(酸性)提取液使之中和,10,000×g、4 ℃离心10 min, 取上清。随后,以试剂盒NAD/NADH Quantification Colorimeteric Kit特异性检测NAD+和NADH,并计算NADH/NAD+。
1.2.6 重组菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm和出发菌C.glutamicumLYS胞内ATP、ADP、AMP的测定[17]
利用0.6 mol/L HClO4(PCA)抽提重组菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm和出发菌C.glutamicumLYS胞内ATP、ADP和AMP。随后,采用HPLC技术对抽提液进行分析,测定胞内ATP、ADP和AMP的浓度。将50 mL细胞样品从培养物中取出,立即在液氮中冷冻60 s,并储存在-20 ℃。通过HPLC测量细胞内ATP的浓度。为了提取ATP,将10 mL 的0.6 mol/L HClO4加入到细胞沉淀中并混合用磁力搅拌器彻底搅拌10 min。将混合物以10 000×g离心10 min以收集上清液。将另外10 mL的0.6 mol/L HClO4加入到沉淀中,充分混合10 min,离心后收集上清液。将两部分上清液在25 mL容量瓶中混合并用0.6 mol/L HClO4补足至25 mL。取10 mL所制备的溶液,并用0.8 mol/L KOH将pH值调节至7.0。在4 ℃保持30 min后,通过过滤从孔中除去晶体KClO4(孔径=0.22 μm),然后在应用HPLC柱之前用磷酸盐缓冲液(pH 7.0)稀释至25 mL。 HPLC分析的进样量为10 μL。使用80%10 mmol/L KH2PO4(pH 7.0)和20%甲醇的混合物作为流动相,流速为1.2 mL/min。紫外检测器的波长设置为260 nm, 柱温控制在25 ℃。
1.2.7 分析方法
菌体浓度测定:将发酵液稀释26倍,测定562 nm处的吸光值。
葡萄糖测定:将发酵液离心(9,000×g,2 min)除去菌体和CaCO3,然后稀释100倍,经生物传感分析仪SBA-40C(山东省科学院生物研究所)测定[18]。
氨基酸含量测定:发酵液中氨基酸含量的测定采用氨基酸自动分析仪[19]。
摇瓶发酵出发菌株C.glutamicumLYS和重组菌株C.glutamicumLYS ΔzwfΔmalEΔicdCg::icdSm,发酵期间定时取样测定发酵液实时葡萄糖含量、菌体量和L-赖氨酸产量并绘制过程曲线。
2 结果与分析
2.1 表达载体和重组菌株的构建
2.1.1 胞内NADPH合成相关基因的敲除载体pK18mobsacB-Δzwf、pK18mobsacB-ΔmalE的构建
E.coliJM109 pK18mobsacB-Δzwf扩大培养后提取质粒分别用BamH I +XbaI和Hind Ⅲ +XbaI双酶切验证基因zwf左右同源臂,从酶切电泳结果图1泳道3、4可以看出,酶切后的左右同源臂片段大小约为900 bp, 与理论大小相符。E.coliJM109 pK18mobsacB-ΔmalE扩大培养后提取质粒分别用Hind Ⅲ +SalI和XbaI +SalI双酶切验证基因malE左右同源臂,从酶切电泳结果图1泳道1、2可以看出,酶切后的左右同源臂片段大小约为900 bp,与理论大小相符。

M-DL5000 Marker;1-质粒pK18mobsacB-ΔmalE Xba I+Sal I双酶切产物,右同源臂;2-质粒pK18mobsacB-ΔmalE HindIII + Sal I双酶切产物,左同源臂;3-质粒pK18mobsacB-Δzwf HindIII + Xba I双酶切产物,右同源臂;4-质粒pK18mobsacB-Δzwf BamH I + Xba I双酶切产物,左同源臂图1 质粒pK18mobsacB-Δzwf、质粒pK18mobsacB-ΔmalE左右同源臂双酶切验证Fig.1 Enzyme digestion of the recombinant plasmid pK18mobsacB-Δzwf and pK18mobsacB-ΔmalE
2.1.2 胞内NADPH合成相关基因的置换载体pK18mobsacB-ΔicdCg::icdSm的构建
E.coliJM109 pK18mobsacB-ΔicdCg::icdSm扩大培养后提取质粒,由于验证时没有合适的酶切位点,只能通过质粒PCR验证基因icdCg左右同源臂和置换基因icdSm,从PCR产物电泳结果图2泳道1可以看出基因icdSm片段大小约为1 600 bp,与理论大小相符,从泳道2、3可以看出,左右同源臂片段大小约为900 bp,与理论大小相符。

M-DL5000 Marker;1-质粒pK18mobsacB-ΔicdCg::icdSm icdSm基因PCR产物;2-质粒pK18mobsacB-ΔicdCg::icdSm右同源臂PCR产物; 3-质粒pK18mobsacB-ΔicdCg::icdSm左同源臂PCR产物图2 质粒pK18mobsacB-ΔicdCg::icdSm各片段PCR验证Fig.2 PCR analysis of the recombinant plasmid pK18mobsacB-ΔicdCg::icdSm
2.1.3 重组菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm的构建
将筛选到的C.glutamicumLYSΔzwf、C.glutamicumLYSΔzwfΔmalE、C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm菌株扩大培养后提取基因组后,分别对应以zwf、malE、icd基因上下游引物进行PC鉴定筛选完成二次同源重组菌株,从PCR产物电泳结果图3泳道1可以看出基因icd置换片段大小约为2 391 bp,与理论大小相符,泳道2、3可以看出基因malE、zwf敲除型残余片段大小约为599、895 bp,与理论大小相符。各PCR产物测序比对结果也与实验设计吻合,最终鉴定正确的转化子命名为C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm。

M-DL5000 Marker;1-基因icd置换型PCR产物;2-基因malE敲除型PCR产物;3-基因zwf敲除型PCR产物图3 重组菌PCR验证Fig.3 PCR analysis of the recombinant strain
2.2 重组菌和出发菌胞内腺嘌呤核苷酸(ATP、ADP和AMP)、吡啶核苷酸(NAD+、NADH、NADP+和NADPH)的变化
为了确定C.glutamicumLYS在敲除zwf、malE基因和替换icd基因后的重组菌是否成功阻断胞内NADPH的合成量,本文对于出发菌和重组菌摇瓶发酵40 h后对其胞内吡啶核苷酸含量进行测定,具体数据如表2。

表2 重组菌和出发菌胞内吡啶核苷酸(NAD+、NADH、NADP+和NADPH)含量Table 2 Contents of intracellular pyridine nucleotides (NAD+, NADH, NADP+ and NADPH) in recombinant and original bacteria
注:a:单位是μmol/g DCW。表3同。
从表2可以看出重组菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm胞内NADH含量由出发菌的1.97 μmol/g DCW增加到2.73 μmol/g DCW,胞内的NADH/NAD+提高了53.84%,NADPH含量降低了89.51%,在该酶的催化下和出发菌株相比不再合成NADPH而合成更多的NDAH,说明自身NADP+-依赖型异柠檬酸脱氢酶基因(icdCg)替换成变形链球菌(Streptococcusmutans)NAD+-依赖型异柠檬酸脱氢酶基因(icdSm),实现了辅酶依赖型的转变;胞内NADPH含量降低至0.15 μmol/g DCW,明显低于出发菌胞内NADPH含量1.43 μmol/g DCW。胞内的NADPH/NADP+降低了93.13%,胞内少量的NADPH,可能是由于阻断了胞内NADPH的合成,NAD(H)/NADP(H)失去平衡后,NAD激酶开始发生作用,使得少量的NADH被NAD激酶催化形成NADPH[24]。谷氨酸棒杆菌体内还有多个涉及NADPH代谢反应没有发现,如呼吸链中NADPH氧化酶的作用[25]。表3可以看出胞内ATP含量降低了13.1%,阻断了胞内NADPH的合成,即敲除了PPP途径和改变了异柠檬酸脱氢酶的辅酶依赖型后不再合成NADPH, NADPH的急剧降低致使经过电子传递链氧化生成的ATP也在一定程度上被削弱,胞内ADP和AMP无法及时转换成ATP,均有一定程度累积,扰乱了胞内腺嘌呤核苷酸平衡。

表3 重组菌和出发菌胞内腺嘌呤核苷酸 (ATP、ADP和AMP)含量Table 3 Contents of intracellular adenine nucleotides (ATP, ADP and AMP) in recombinant and original bacteria
2.3 阻断胞内NADPH合成途径对葡萄糖消耗速率和菌体生长的影响
由图4可知出发菌和重组菌的生长趋势基本保持一致,但重组菌的生长较为缓慢,生长速率偏低,在相等OD562相同的接种量下,菌体延滞期基本相同,而菌体的对数生长期延长约4 h。由图5可知在发酵过程的12~24 h,主要在菌体的对数生长期重组菌的葡萄糖代谢能力明显弱于出发菌株。
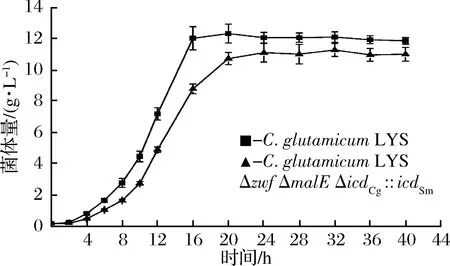
图4 出发菌和重组菌株菌体生长曲线Fig.4 Growth curve of the oringinal and recombinant strains

图5 出发菌和重组菌株葡萄糖消耗速率Fig.5 Glucose consumption rate of the oringinal and recombinant strains
由于戊糖磷酸途径是糖代谢的第二条重要途径同样也是细胞产生还原力(NADPH)的主要途径,敲除了合成NADPH的PPP途径,胞内NADPH锐减,减缓胞内各种需要NADPH的合成反应,如氨基酸、脂类及核苷酸等细胞组成物质的合成,对细胞正常生长和代谢有重要影响,发酵结束后重组菌菌体量与出发菌相比降低了4.36%。阻断了葡萄糖进入PPP途径,葡萄糖代谢途径减少致使葡萄糖的代谢减缓。胞内的NADH/NAD+提高了53.84%,更高的NADH/NAD+比率不利于糖酵解的进行,致使重组菌的葡萄糖代谢能力进一步削弱。胞内NAD激酶发生作用,形成少量的NADPH优先供应细胞生长,致使阻断胞内L-赖氨酸合成通路中NADPH的合成,对菌体生长影响相对较小,而对L-赖氨酸产量等代谢产物影响较大。菌体生长也因葡萄糖代谢减缓而受到一定程度的影响。而NADPH也会影响胞内微环境,如NAD(H/+)状态和ATP含量,由于ATP可调节胞内pHi,提高细胞对酸胁迫的适应力,NADPH可通过NADPH氧化酶阻止胞内羟基等活性氧簇(ROS)的形成,提高细胞对氧胁迫的适应力[26]。而阻断了NADPH合成途径导致胞内微环境扰乱,有可能间接性的影响到菌体生长和葡萄糖代谢。
2.4 阻断胞内NADPH合成途径对L-赖氨酸合成的影响
由图6可知,40 h发酵结束后出发菌C.glutamicumLYS发酵液中L-赖氨酸含量为6.5 g/L,重组菌C.glutamicumLYS ΔzwfΔmalEΔicdCg::icdSm,L-赖氨酸产量为0.8 g/L,与出发菌相比,阻断胞内NADPH合成途径胞内的NADPH降低了89.51%,L-赖氨酸产量降低了87.69%。

图6 摇瓶培养条件下出发菌和重组菌株L-赖氨酸产量Fig.6 L-lysine yield of the oringinal and recombinant strains in shake flask culture
敲除了葡萄糖-6-磷酸脱氢酶编码基因使得PPP途径失活“C”通量减少,TCA循环“C”通量增加,苹果酸酶编码基因敲除和异柠檬酸脱氢酶由于基因置换导致辅酶依赖型的改变使得胞内NADPH含量降低至0.15 μmol/g DCW。在发酵12 h后,出发菌和重组菌的菌体量开始出现较大的差距,同时L-赖氨酸含量也开始出现很明显的差距,结果表明此时由于重组菌无法满足菌体生长和L-赖氨酸合成所需的NADPH,导致菌体生长缓慢,菌体量减少,并且在L-赖氨酸合成途径中谷草转氨酶、天冬氨酸半醛脱氢酶、二氢吡啶二羧酸还原酶、内消旋二氨基庚二酸脱氢酶或琥珀酰-氨基-吡咯酮转氨酶需要以NADPH为辅酶,合成1分子L-赖氨酸需要4分子的NADPH,NADPH的严重缺乏,加剧了L-赖氨酸的产量的降低。大量研究表明,L-赖氨酸产量的增加与增加PPP途径“C”通量和减少TCA循环“C”通量密切相关。
2.5 阻断胞内NADPH合成途径对副产物合成的影响
很多氨基酸的合成都依赖于NADPH,为了进一步研究阻断胞内NADPH合成途径对发酵副产物如有机酸和其他氨基酸的影响,发酵结束后测定发酵液中有机酸和副产物氨基酸含量。
由表4可以看出由于阻断了胞内NADPH合成途径出发菌C.glutamicumLYS和重组菌C.glutamicumLYS ΔzwfΔmalEΔicdCg::icdSm发酵液中的副产物氨基酸明显降低,缬氨酸、亮氨酸、异亮氨酸、苏氨酸、谷氨酸较出发菌株分别降低了76.94%、79.22%、 83.09%、80.87%、62.25%,缬氨酸、亮氨酸、异亮氨酸、苏氨酸、谷氨酸的合成都需要提供辅因子NADPH,致使合成产量都明显降低,而由于合成谷氨酸所需要的辅因子NADPH相对较低,对其生物合成的影响相比其他氨基酸而言相对较低,而赖氨酸合成需要更多的辅因子NADPH,致使其产量降低的最为明显。

表4 重组菌和出发菌发酵液中副产物氨基酸和有机酸含量Table 4 The concentration of amino acid and organic acid in fermentation of recombinant and original bacteria
而重组菌较低的菌体量对发酵过程中各种氨基酸副产物的积累也有一定程度的削弱。发酵液中的有机酸测定结果表明,发酵液中的丙酮酸和乳酸含量与出发菌相比分别增高了41.55%、15.73%,是由于阻断了胞内NADPH合成途径后,极大削弱了菌株内以NADPH为辅酶的氨基酸合成,如以丙酮酸为前体,合成的丙氨酸族氨基酸,和经过丙酮酸流向三羧酸循环后以草酰乙酸为前体合成的天冬氨酸族氨基酸,使得丙酮酸等代谢途径上游的中间产物得以累积。
3 结论
该研究中对L-赖氨酸产生菌C.glutamicumLYS进行基因工程改造,构建了重组自杀型质粒pK18mobsacB-Δzwf、pK18mobsacB-ΔmalE用于敲除葡萄糖-6-磷酸脱氢酶编码基因zwf和苹果酸酶编码基因malE,克隆来源于Streptococcusmutans的NADP+-依赖型异柠檬酸脱氢酶(编码基因icdCg)通过重组自杀型质粒pK18mobsacB-ΔicdCg::icdSm替换谷氨酸棒杆菌NAD+-依赖型异柠檬酸脱氢酶(编码基因icdSm),依次将重组质粒电转化至C.glutamicumLYS,筛选并获得完成二次同源重组的菌株C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm,该重组菌阻断了谷氨酸棒杆菌中NADPH合成的相关途径。由于异柠檬酸脱氢酶是三羧酸循环中将柠檬酸催化生成异柠檬酸,通过基因置换以改变其辅因子依赖型,本研究中用来源于Streptococcusmutans的异柠檬酸脱氢酶替换谷氨酸棒杆菌原有的异柠檬酸脱氢酶。对出发菌和重组菌进行摇瓶发酵实验,对两种不同的辅因子NADPH水平下胞内氧化还原辅因子(NAD+、NADH、NADP+和NADPH)和能量辅因子(ATP、ADP和AMP)进行分析和比较,NADPH含量较出发菌降低了89.51%,NADPH/NADP+降低了93.13%,NADH含量提高了38.58%,NADH/NAD+提高了53.84%,ATP含量降低了13.1%。重组菌的葡萄糖代谢能力明显弱于出发菌株,菌体的生长也相对缓慢,进入稳定期的时间延长,最终菌体量与出发菌相比降低了4.36%。L-赖氨酸产量降低了87.69%, 缬氨酸、亮氨酸、异亮氨酸、苏氨酸、谷氨酸合成量也都明显降低,而在胞内积累了大量副产物,如丙酮酸、乳酸等。当阻断胞内NADPH的合成途径时,胞内微环境如氧化还原水平等受到扰动,此时菌体生长受到影响,而对L-赖氨酸产量的改变尤为明显。为进一步解析辅因子NADPH调控L-赖氨酸产生菌胞内微环境的生理机制奠定基础。
猜你喜欢
家庭科学·新健康(2021年5期)2021-06-21
科学大众(2021年10期)2021-05-20
中国兽医杂志(2019年10期)2019-05-20
心肺血管病杂志(2018年11期)2018-12-18
中成药(2017年12期)2018-01-19
中国畜牧业(2016年12期)2016-02-17
中国医药生物技术(2015年4期)2015-12-26
安徽医科大学学报(2015年9期)2015-12-16
中国畜牧业(2014年5期)2014-12-20
中国人兽共患病学报(2014年1期)2014-04-02
