
甲状腺癌患者预后风险模型的构建
2019-11-15 06:12何东生曾先捷
实用癌症杂志 2019年11期
何东生 曾先捷
甲状腺癌(thyroid cancer,TC) 作为全球发病率增速最快的肿瘤,是内分泌系统中最常见的恶性肿瘤[1]。虽然甲状腺癌患者5年生存率相对其他恶性肿瘤患者较高,但是在过去的十年里,甲状腺癌患者的死亡率仍持续增加[2-3]。2017年,全球甲状腺癌新发病例数占所有肿瘤新发病例的3.4%,在女性肿瘤中发病率上升到第五位[4-5]。甲状腺癌已经成为1个不容忽视的全球性问题[6]。若能进一步明确甲状腺癌的独立预后因子,对判断甲状腺癌患者的预后、寻找新的治疗靶点和提高患者总体生存率将会起到至关重要的作用。目前,预测甲状腺癌的生存预后主要通过患者的临床-病理特征进行判断[7]。然而,随着对甲状腺癌分子机制研究的深入,通过基因预测甲状腺患者预后正逐步受到重视[8]。本研究对TCGA数据库中甲状腺癌患者的mRNAs测序数据及相应的生存数据进行分析。在对与甲状腺癌生存预后显著相关的mRNAs进行cox回归分析之后,筛选出6个潜在的独立预后因子并构建预测甲状腺癌患者预后风险的多因素cox回归模型,为甲状腺癌患者的治疗及改善预后提供参考依据。
1 资料与方法
1.1 数据资料收集
下载TCGA数据库(https://portal.gdc.cancer.gov/)中人类甲状腺癌的RNA-Seq数据和507例患者临床信息。RNA-Seq数据中包括58例正常甲状腺样本和510例甲状腺癌样本的测序数据。通过R软件(https://www.r-project.org/)对所下载数据进行整理。
1.2 差异基因的筛选
利用edgeR函数包[9]对甲状腺正常样本和癌样本进行分析,筛选出差异表达的mRNA。通过除异均值化M值法(trimmed mean of M values,TNM)对RNAs的表达量进行标准化处理后[10],采用错误发现率(false discocery rate,FDR)对RNAs的显著性进行校正[11]。差异表达的RNAs的cut-off值设定为:①FDR≤0.01;②∣log2fold change (FC)∣≥2。
1.3 单因素和多因素Cox回归分析
排除5例缺失mRNA测序数据的甲状腺癌患者资料,将502例甲状腺癌患者的生存数据与相应的差异表达的mRNA测序数据相合并。利用survival函数包对差异表达的mRNA和甲状腺癌患者的生存预后之间的关系进行单因素Cox回归分析[12]。与甲状腺癌患者生存预后显著相关的mRNAs(P<0.001)被纳入多因素cox回归分析以进一步检验这些基因判断甲状腺癌患者预后的价值。通过以上分析,构建判断甲状腺癌患者预后风险的cox回归模型:风险值 (Risk Score)=expmRNA1×βmRNA1+expmRNA2×βmRNA2+……+expmRNAn×βmRNAn(exp:表达水平;β:多因素cox回归分析的回归系数)[13]。
1.4 甲状腺癌患者的高低风险划分以及ROC 曲线的绘制
根据预测甲状腺癌患者预后风险的cox回归模型,计算502个甲状腺癌患者的风险值。以中位风险值为界,将患者分为高风险组和低风险组并进行Kaplan-Meier生存曲线的绘制和log-rank分析。使用survivalROC函数包绘制ROC (receiver operating characteristic)曲线[14],判断通过cox回归模型预测甲状腺癌患者5年生存率的准确性。
2 结果
2.1 差异基因的筛选
对TCGA数据库中58个正常甲状腺癌样本和510个甲状腺癌样本的测序数据进行分析后,发现17651个基因中的1097mRNAs差异表达(233个下调,864上调)。
2.2 单因素cox和多因素cox回归分析
通过survival函数包进行单因素cox回归分析,对甲状腺癌中1097个差异表达的mRNAs与患者生存预后之间的关系进行探索。如表1所示,11个差异表达的mRNA与甲状腺癌患者的生存预后显著相关(P<0.001)。通过多因素cox回归分析对11个基因预测患者预后的能力进行进一步检验后,我们发现RIPPLY3、PCOLCE2、FAM111B、ZSCAN4、SALL3、DLK1六个基因具有充当独立预后因子的功能。判断甲状腺癌患者预后风险值的多因素cox回归模型为:风险值=(RIPPLY3×-0.6194) + (PCOLCE2×0.5299) + (FAM111B×-0.3838) + (ZSCAN4×-0.4329) + (SALL3×0.3508) + (DLK1×0.2216)。RIPPLY3、FAM111B、ZSCAN4在甲状腺癌患者低风险组的表达量高于高风险组,PCOLCE2、SALL3、DLK1在甲状腺癌低风险组的表达量低于高风险组。

表1 与甲状腺癌患者生存预后显著相关的差异表达基因
2.3 甲状腺癌患者的高低风险划分以及ROC曲线的绘制
根据判断甲状腺癌患者预后风险值的多因素cox回归模型,分别计算502个甲状腺癌患者的预后风险值,以中位数为界,将患者分为高风险组和低风险组。Kaplan-Meier生存分析结果显示甲状腺癌高风险患者与低风险患者的生存率之间存在显著差异,高风险组甲状腺癌患者的生存预后显著劣于低风险组甲状腺癌患者,见图1。高风险组甲状腺癌患者的五年生存率为84.7%,低风险组甲状腺癌患者的生存率为100%。ROC曲线下面积为0.955(图2),确认了基于多因素cox回归模型预测甲状腺癌患者预后风险的准确性。
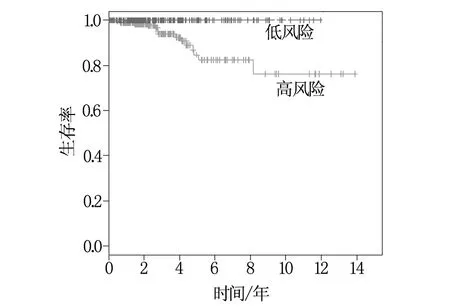
图1 甲状腺癌患者高风险组和低风险组的Kaplan-Meier生存分析

图2 ROC曲线判断患者预后的准确性
3 讨论
甲状腺癌作为内分泌系统最常见的恶性肿瘤,在形态学特征和预后方面存在着巨大的异质性[6]。由于甲状腺癌复发率的增高,导致甲状腺癌患者不可治愈率以及死亡率有所增加[15]。目前,预测肿瘤患者生存的预后主要依靠患者临床特征,如年龄,性别,肿瘤位置、大小、分期、转移等[7-8]。然而,随着对甲状腺癌分子层面研究的深入,这一观点正不断受到质疑[8]。本研究通过对甲状腺癌中异常表达的基因进行探索,筛选出与患者总体生存预后显著相关的核心基因,从基因表达方面对甲状腺癌患者的预后风险进行预测。
近几年,微阵列和高通量测序技术的发展在揭示癌症发生发展过程中基因的异常表达和识别与癌症诊断和预后相关的生物标记物方面起到至关重要的作用[16]。本研究对TCGA数据库中甲状腺癌患者的基因表达谱及相应的临床信息进行综合性分析。通过对58例正常甲状腺样本和510例甲状腺癌样本的测序数据进行分析,我们筛选出了233个下调和864上调的mRNAs。在对1097个差异表达的mRNAs进行单因素cox回归分析筛选出与甲状腺癌患者预后显著相关的mRNAs之后,将其纳入多因素cox回归分析并筛选出6个甲状腺癌的独立预后因子:RIPPLY3、PCOLCE2、FAM111B、ZSCAN4、SALL3和DLK1。基于这6个基因构建具有预测患者预后风险功能的多因素cox回归模型。RIPPLY3、FAM111B、ZSCAN4基因的多因素cox回归系数均小于0,提示这三个基因高表达患者的预后会优于低表达的患者。PCOLCE2、SALL3、DLK1基因的多因素cox回归系数均大于0,提示这3个基因低表达患者的预后会优于高表达的患者。在对502例甲状腺癌患者进行风险值的评分后,将患者划分为高风险组和低风险组并进行Kaplan-Meier生存分析,结果证明高风险组甲状腺癌患者的总体生存率显著劣于低风险组患者。ROC曲线验证了通过6个甲状腺癌独立预后因子判断甲状腺癌患者风险值的可靠性。通过预测甲状腺癌患者的预后风险值,可以为甲状腺癌患者进行个性化治疗提供参考,从而降低患者死亡率,改善患者预后。本研究的不足之处在于所有数据均来自TCGA数据库提供的甲状腺癌mRNA测序数据,可能无法完全代表其蛋白水平表达的情况。在后续的实验中,应该结合免疫组化、蛋白印记以及细胞功能等试验进一步讨论和验证筛选出来的6个甲状腺癌独立预后因子在甲状腺癌中的表达情况以及在预测患者生存预后方面所起的作用。虽然6个基因在甲状腺癌的蛋白表达水平以及功能有待进一步研究,但在其他癌种中已有相关报道。ZSCAN4通过调节端粒的长度维持基因组稳定,在肿瘤细胞以及胚胎干细胞中,ZSCAN4在延伸端粒长度方面起到重要调节作用[17-18];PCOLCE2在卵巢癌中起到了促癌因子的作用,不利于患者临床预后;FAM111B的低表达被认为与子宫颈癌患者的远处转移有关[19-20]。此外,DLK1被认为可作为胚胎癌的治疗靶点[21]。
总之,本研究通过对甲状腺癌患者的基因表达谱及其临床信息进行综合分析,发现了6个最有可能具有预测患者生存预后功能的独立因素并构建预后风险预测模型,对甲状腺癌患者的诊断和治疗和改善预后提供了参考依据。
猜你喜欢
核安全(2022年2期)2022-05-05
铁道建筑技术(2021年4期)2021-07-21
国际放射医学核医学杂志(2021年10期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
祝您健康·文摘版(2020年7期)2020-07-13
中国生殖健康(2019年2期)2019-08-23
世界知识(2018年19期)2018-11-21
老年教育(老年大学)(2018年12期)2018-01-28
北方文学·下旬(2017年4期)2017-05-21
中国卫生标准管理(2015年2期)2016-01-15
