
一种新型手性胍催化剂的合成和表征*
2020-03-08 09:06胡家栋
化学与粘合 2020年6期
胡家栋,文 雯,陈 乐
(1.杨凌职业技术学院 药物与化工分院,陕西 杨凌 712100;2.西北农林科技大学 化学与药学院,陕西 杨凌 712100)
前言
胍基广泛存在于多种具有重要生物活性的天然产物中[1,2],为天然产物结构和功能的多样性提供了物质基础。比如,具有抗病毒、抗真菌和抗HIV 活性的海洋胍类生物碱ptilomycalin A[3]和crambescidins以及多种酶活性位点上精氨酸侧链中都含有胍基(图1a)。同时,由于其共轭酸的稳定性[4],胍是一种有机强碱,能够催化Strecker 反应[5]、Michael 加成反应[6]、Henry 反应[7]和Mannich 反应[8]等多种碱介导的有机反应。并且,胍基能够在较大的pH 值范围内被质子化而保持正电性[9],易与负离子以氢键形式形成紧密离子对,从而有效地维持了负离子的立体构型[10~14],这为开发手性胍催化剂用于不对称催化反应提供了理论基础。
手性胍催化剂是除了L-脯氨酸及其衍生物、金鸡纳碱及其衍生物、手性磷酸、手性硫脲和氮杂环卡宾等以外的另一类重要的有机小分子催化剂,已经被广泛地应用于不对称催化领域[10~21],在重要医药、农药中间体的不对称合成当中起着至关重要的作用[12,14]。从结构上看,胍基的3 个氮原子上至少可以引入5 个手性侧链(图1b)。根据手性侧链连接方式的不同,可以将手性胍催化剂分为双环手性胍催化剂、单环手性胍催化剂和开环手性胍催化剂三种类型[12]。目前,文献报道的开环手性胍催化剂以手性胍-硫脲[22~24]和氨基酸衍生的手性胍[25,26]为主,所催化的反应类型也较少。因此,开发一种结构新颖的新型开环手性胍催化剂,对于手性胍催化不对称反应的研究显得十分必要。
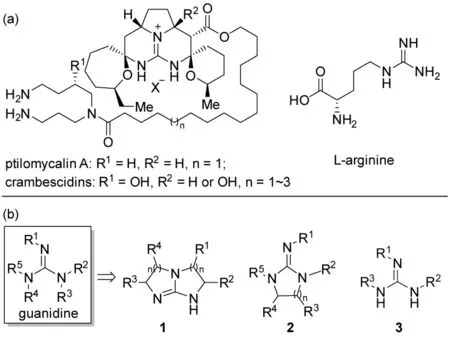
图1 含胍基的天然产物及胍类催化剂的分类Fig.1 The natural products containing the guanidine and the categorization of guanidine catalyst
本文介绍了一种以商业可得的S-1,1’-联-2-萘酚为原料,经三氟甲磺酸酯化、Kumada 偶联、自由基溴代反应和N-烷基化四步转化,以53.4%的总收率合成一种新型的开环手性胍催化剂的方法,并通过核磁、质谱和X-ray 单晶衍射实验对其化学结构进行了表征。
1 实验部分
1.1 主要原料与试剂
S-1,1'-联-2-萘酚,三氟甲磺酸酐,三乙胺,甲基溴化镁,1,3-双(二苯基膦丙烷)二氯化镍,N-溴代丁二酰亚胺,偶氮二异丁腈,胍,二氯甲烷,乙醚,乙酸乙酯,四氯化碳,乙醇,四氢呋喃,硅藻土,饱和食盐水和无水硫酸钠等。
1.2 仪器与设备
IKA C-MAG HS7 型加热磁力搅拌器,IKA RV8型旋转蒸发仪,顾村ZF-II 型四用紫外分析仪,Bruker AMX-500 型核磁共振仪,Finnigan MAT 型质谱仪,Bruker Smart Apex CCD 单晶衍射仪等。
1.3 新型手性胍催化剂的合成
新型手性胍催化剂8 可以由两分子二溴化合物7 与一分子胍的乙醇溶液经N-烷基化反应以68%的收率得到。而关键底物二溴化合物7 又可以从商业可得的S-1,1’-联-2-萘酚4 出发,通过改进的文献方法[27]经三氟甲磺酸酯化、Kumada 偶联和自由基溴代反应三步转化以高达78.4%的总收率得到。
合成路线如图2 所示,具体的合成实验操作列举如下:

图2 新型手性胍催化剂8 的合成Fig.2 The synthesis of novel chiral guanidine catalyst 8
中间体5 的合成(三氟甲磺酸酯化):取一250mL的圆底烧瓶,依次加入3.00g 的S-1,1'-联-2-萘酚和120mL 二氯甲烷,并用冰浴将体系降至0℃。逐滴加入2.06mL 三乙胺并搅拌30min。接着,逐滴加入3.70mL 三氟甲磺酸酐并回流搅拌18h。TLC 监测原料4 完全消耗后,将体系冷却至室温并用二氯甲烷稀释。在冰浴条件下,逐滴加入1.0M 的盐酸10mL 淬灭反应。用二氯甲烷萃取,饱和NaHCO3溶液中和酸性,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用硅胶柱层析分离得白色粉末状的中间体5 共5.75g,收率99%。
中间体6 的合成(Kumada 偶联):取一50mL 的圆底烧瓶,依次加入1.80g 双三氟甲磺酸酯5,177mg的1,3-双(二苯基膦丙烷)二氯化镍和12mL 乙醚,并用冰浴将体系降至0℃。逐滴加入4.35mL 甲基溴化镁溶液(3.0Min Hexane)并升温回流18h。TLC 监测原料5 完全消耗后,将体系冷却至室温并用乙酸乙酯稀释。在冰浴条件下,逐滴加入1.0M 的盐酸10mL 淬灭反应。用乙酸乙酯萃取,饱和NaHCO3溶液洗涤,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用硅胶柱层析分离得无色油状的中间体6 共913mg,收率99%。
中间体7 的合成(溴代反应):取一50mL 的圆底烧瓶,依次加入0.67g 的化合物6,0.91g 的N-溴代丁二酰亚胺,19.4mg 的偶氮二异丁腈和20 mL 经脱气处理的四氯化碳。将体系升温至回流搅拌24h。TLC 监测原料6 完全消耗后,将体系冷却至室温并用硅藻土过滤,滤饼用二氯甲烷洗涤。接着,将有机相用饱和NaHCO3溶液中和,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用硅胶柱层析分离得白色固体的二溴化合物7 共0.82g,收率80%。
新型手性胍8 的合成:取一50mL 的圆底烧瓶,依次加入5.00mL 胍的乙醇溶液(1.0M in EtOH),440mg 二溴化合物7 和15mL 四氢呋喃。在室温条件下搅拌12h。TLC 监测原料7 完全消耗后,用1.0M的盐酸10mL 淬灭反应。用二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用硅胶柱层析分离得胍的盐酸盐221mg,收率68%。然后,用2.0M的氢氧化钾水溶液中和盐酸盐,用二氯甲烷萃取并旋干溶剂得白色固体。在二氯甲烷/甲醇溶液中缓慢挥去溶剂培养出单晶。
2 结果与讨论
2.1 中间体5、6 和7 的数据表征
中间体5、6 和7 的化学结构通过核磁氢、碳谱实验进行确证,并与文献[27]报道数据进行了逐一比对从而确认了其结构。以上三个化合物的氢、碳谱数据分别解析如下:
中间体5 的数据表征:1H NMR(500MHz,CDCl3):δ 8.14(d,J=9.0Hz,2H),8.01(d,J=8.0Hz,2H),7.61(d,J=8.5Hz,2H),7.57~7.60(m,2H),7.39~7.43(m,2H),7.25(d,J=7.0Hz,2H).13C NMR(125MHz,CDCl3):δ 178.1,142.2,136.3,131.2,129.3,127.0,126.2,43.1,41.4,41.9,26.2,22.5.
中间体6 的数据表征:1H NMR(500MHz,CDCl3):δ7.89(t,J=8.0Hz,4H),7.51(d,J=8.5Hz,2H),7.38~7.41(m,2H),7.19~7.22(m,2H),7.04(d,J=8.5Hz,2H),2.03(s,6H).13C NMR(125MHz,CDCl3):δ 178.1,142.2,136.3,131.2,129.3,127.0,126.2,43.1,41.4,41.9,26.2,22.5.
中间体7 的数据表征:1H NMR(500MHz,CDCl3):δ 8.04(d,J=8.5Hz,2H),7.94(d,J=8.0Hz,2H),7.77(d,J=8.5Hz,2H),7.50(t,J=7.5Hz,2H),7.28(t,J=7.5Hz,2H),7.11(d,J=8.0Hz,2H),4.28(s,4H).13C NMR(125MHz,CDCl3):δ 178.1,142.2,136.3,131.2,129.3,127.0,126.2,43.1,41.4,41.9,26.2,22.5.
2.2 化合物8 的核磁数据

图3 新型手性胍催化剂8 的1H NMR 谱图Fig.3 The 1H NMR spectrum of novel chiral guanidine catalyst 8
新型手性胍催化剂8 的1H NMR 谱图数据如图3 所示。数据解析如下:1H NMR(300MHz,CDCl3)δ 8.06~7.92(m,8H),7.55~7.37(m,12H),7.31~7.25(m,4H),4.41(d,J=12.6Hz,4H),4.14(d,J=12.6Hz,4H)。
从氢谱上看,在8.06~7.25 范围内共有24 个芳香氢,与两个联二萘环上的氢数目相匹配;在4.41和4.14 处分别有峰型为双峰的4 个氢,且其耦合常数相等,均为12.6Hz,与四个苄位的8 个氢相匹配,初步证明该化合物的结构是手性胍8。
2.3 化合物8 的质谱数据
新型手性胍催化剂8 的质谱数据如图4 所示。数据解析如下:MS(ESI)base peak found:m/z 616.70[M+H]+.
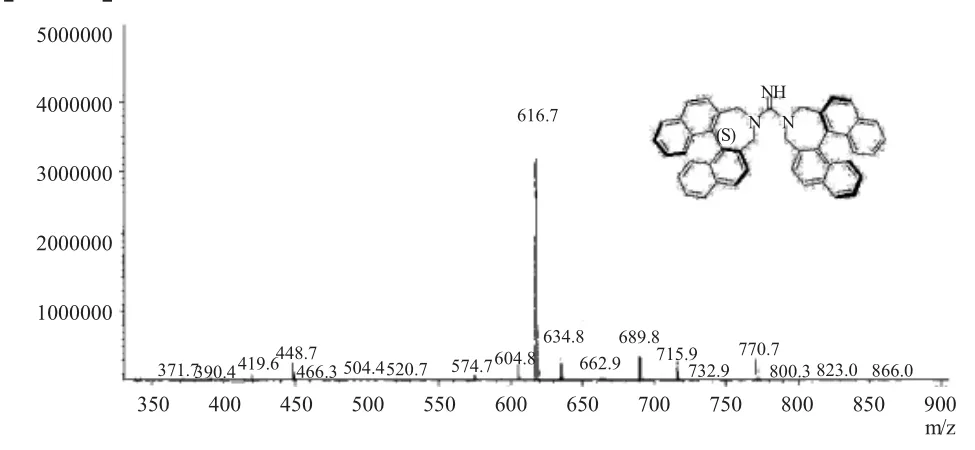
图4 新型手性胍催化剂8 的质谱图Fig.4 The MS spectrum of novel chiral guanidine catalyst 8
从质谱上看,以ESI 电离方式得到的分子离子峰基峰m/z 在616.70 处有信号,而手性胍8 的m/z 为615.28,说明检测到的是[M+H]+信号。质谱数据进一步佐证了该化合物是手性胍8。
2.4 化合物8 的晶体结构图
化合物8 的单晶在Bruker Smart Apex CCD 单晶衍射仪上测试,用经石墨单色器单色化的Mo-Kα射线(λ=0.71069Å)照射,用SHELXL-2015/1 程序以直接法解出。得到化合物8 的晶体结构如图5 所示,最终确证了所得新型开环手性胍8 的空间结构。
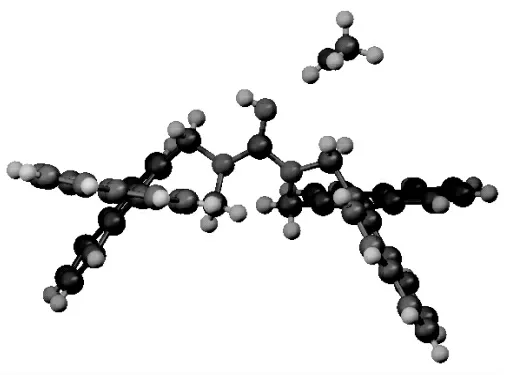
图5 新型手性胍催化剂8 的晶体结构图Fig.5 The ORTEP of novel chiral guanidine catalyst
在这个单晶结构中,两个萘环的二面夹角分别为60.19°、60.24°。在化合物8 的单晶中还带有一个甲醇分子,胍基的氮原子和甲醇分子的羟基之间存在氢键作用。
3 结论
本项目以商业可得的S-1,1’-联-2-萘酚为原料,通过对文献已有合成二溴化合物7 方法的改进,经三氟甲磺酸酯化、Kumada 偶联和自由基溴代反应三步转化,以高达78.4%的总收率合成了重要中间体7。接着,通过二溴化合物7 与胍的乙醇溶液的N-烷基化反应以68%的收率合成了一种新型的开环手性胍催化剂,并在二氯甲烷/甲醇溶液体系中成功培养了化合物的单晶,通过核磁、质谱和X-ray 单晶衍射实验对催化剂的结构进行了确证。这一新型开环手性胍催化剂的合成与结构表征为后续对这类催化剂催化的不对称反应研究奠定了物质基础。目前,对这一新型催化剂的进一步应用开发及应用于工业生产改造的研究工作正在积极的进行当中。
猜你喜欢
佳木斯大学学报(自然科学版)(2022年1期)2022-11-25
分子催化(2022年1期)2022-11-02
科学导报(2022年41期)2022-07-13
中国药学药品知识仓库(2022年10期)2022-05-29
中国典型病例大全(2022年9期)2022-04-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
中国药科大学学报(2020年4期)2020-09-14
湖南大学学报·自然科学版(2020年2期)2020-04-17
健康必读·下旬刊(2019年5期)2019-06-03
江苏农业科学(2017年20期)2017-11-30
