
ZFN, TALEN和CRISPR/Cas9在小鼠Rosa26基因定点整合外源基因的效率比较*
2020-04-16 13:22刘小凤刘蔚聂宇丛佩清刘小红陈瑶生何祖勇
中山大学学报(自然科学版)(中英文) 2020年2期
刘小凤,刘蔚,聂宇,丛佩清,刘小红,陈瑶生,何祖勇
(中山大学生命科学学院/有害生物控制与资源利用国家重点实验室,广东 广州 510006)
锌指核酸酶(zinc-finger nuclease,ZFN)技术、类转录激活效应因子核酸酶 (transcription activator-like effector nuclease,TALEN)技术和成簇规律间隔短回文重复(clustered regulatory interspaced short palindromic repeat,CRISPR)/Cas9技术是近年来新发展的三种基因组编辑技术,它们均是通过引导核酸内切酶切割DNA靶位点序列,形成DNA双链断裂(DNA double-strand break,DSB),从而诱导细胞进行DNA损伤修复。细胞一般通过非同源末端连接(non-homologous end joining,NHEJ)或同源重组(homology-directed repair,HDR)两种不同的DNA修复途径来实现基因组编辑,包括基因敲除(knockout)、基因定点突变及外源基因的定点整合(knockin)等。
ZFN由锌指蛋白(ZFP)和IIS型限制性内切酶FokⅠ融合构成。3~5个串联的ZFP构成ZFN的DNA结合结构域,可与DNA靶位点特异性结合,然后引导FokⅠ蛋白二聚体发挥DNA切割功能[1- 2]。TALEN技术与ZFN技术类似,是通过类转录激活效应因子(TALE)与DNA靶位点特异结合,诱导FokⅠ蛋白二聚体在靶位点进行切割[3]。而CRISPR/Cas9技术则是通过人工合成的单链引导RNA(single strand guide RNA, sgRNA)序列与靶位点特异性结合,引导Cas9蛋白进行DNA双链切割[4]。近年来,这三种基因组编辑技术以高效、精准的特点在基因敲除方面得到了广泛应用,但是对于定点整合外源基因,三者达到的效率参差不齐,且目前尚未见在同一位点上对这三种基因组编辑技术介导的外源DNA片段整合效率进行系统比较的研究报道。因此本研究设计了靶向小鼠Rosa26基因同一个位点的ZFN、TALEN和CRISPR/Cas9,首先在C2C12细胞中比较了三者的靶向切割效率,然后构建了一个含有黑色素生成相关的酪氨酸酶基因Tyr和红色荧光蛋白报告基因DsRed的无启动子同源重组质粒供体pTyr-2A-DsRed,在基因编辑工具的介导下,只有正确整合到小鼠Rosa26基因的供体载体才能启动Tyr及DsRed基因的表达,通过对比转染后的细胞红色荧光强度,本研究系统比较了三种基因组编辑工具介导外源DNA定点整合的效率,结果发现CRISPR/Cas9的效率最高,在此基础上,利用CRISPR/Cas9将供体整合到小鼠胚胎干细胞中,筛选单细胞克隆进行囊胚腔注射和胚胎移植,获得一只存活的嵌合体小鼠,表现出白毛中夹杂黑毛的表型,这表明整合到小鼠Rosa26的Tyr基因可以正常表达。
1 材料和方法
1.1 材料
C2C12细胞由本实验室保存,小鼠胚胎干细胞由中山大学实验动物中心提供,昆明小鼠(Kunming mice, KM)来自中山大学实验动物中心,pX458质粒(Cas9/sgRNA共表达质粒)购自Addgene公司。PCR引物及载体构建相关的oligo序列由上海生工生物技术公司合成。
1.2 方法
1.2.1Rosa26基因靶位点选择 应用ZiFiT在线分析软件(http://zifit.partners.org/ZiFiT/)对小鼠Rosa26基因内含子1的序列进行分析,设计1对ZFN和2对TALEN;应用CRISPR DESIGN (http://crispr.mit.edu/)设计一条gRNA,使三者的切割位点几乎重叠(图1:A)。
1.2.2 ZFN、TALEN与CRISPR/Cas9表达载体构建
1) ZFN表达质粒构建
ZFN与Rosa26内含子的结合区域如图1:A所示,两段ZFN结合序列中间的序列为切割位点。ZFN质粒由SIGMA公司合成。
2) TALEN表达质粒构建
TALEN与Rosa26内含子的结合区域如图1:A所示,TALEN质粒采用本实验室前期使用的“Golden Gate”方法构建[5]。
3) CRISPR/Cas9表达质粒构建
pX458载体可同时表达sgRNA、Cas9蛋白以及EGFP荧光蛋白,通过BbsⅠ酶切可将gRNA序列连进载体。合成gRNA正负单链时,在5′端加上与BbsⅠ内切酶开pX458载体后的粘性末端互补的序列(表1),将合成好的gRNA正负单链粉末稀释至100 μmol/L,进行gRNA的磷酸化和退火,使之形成具有粘性末端的双链DNA短片段。将退火并磷酸化的gRNA双链短片段和pX458载体通过BbsⅠ内切酶和T4连接酶进行酶切和连接反应,接着进行转化,挑取单克隆,测序验证CRISPR/Cas9表达质粒是否构建成功。

图1 ZFN、TALEN、CRISPR/Cas9 靶向切割效率鉴定

表1 靶向小鼠Rosa26基因的gRNA oligo序列
1.2.3 同源重组质粒供体构建 在小鼠中,黑色素沉积的类型与数量差异造成了不同的毛色表型。动物的黑色素分为两类:真黑色素(eumelanin)与褐黑色素(pheomelanin),这两种黑色素的合成均依赖于酪氨酸的氧化,而酪氨酸酶(Tyr)是催化酪氨酸氧化的关键酶[6]。当外源Tyr基因整合进白毛色小鼠中,成功整合外源Tyr基因的白色小鼠会产生黑色素沉积,产生不同于对照组的毛色表型[7]。因此当构建基因敲入小鼠时,Tyr可以作为一个遗传筛选标记,使我们可以通过毛色来快速、高效地鉴别Knockin小鼠。Parikh团队在2015年用CRISPR/Cas9技术插入了Tyr基因,并成功得到了毛色改变的嵌合体小鼠[8]。
为方便、快捷地获得Tyr基因整合的小鼠,我们构建了pTyr-2A-DsRed同源重组质粒供体(图2: A),供体包含位于两侧的同源臂和位于中间的外源插入序列。研究表明,同源臂长度为1 kb左右时具有最优的同源重组效率[9]。我们所设计的5′同源臂序列长度为921 bp,3′同源臂序列长度873 bp。插入序列包括了需整合的Tyr基因序列与红色荧光报告基因(DsRed)序列,此外,还有一个剪接受体位点(splicing acceptor, SA),该受体位点的存在使插入序列被整合到基因组后,在转录后剪接时可与5′端外显子连接,保证插入序列可以正常翻译[10-11]。
1.2.4 细胞转染 选用15代以内的细胞,胰酶消化后,1 600 r/min离心4 min,吸掉上清,加入1 mL PBS洗涤并用细胞计数仪计算细胞数量。计数后,将PBS细胞悬液1 600 r/min离心4 min,吸掉上清,按每1×106细胞加入100 μL细胞悬浮液R,吹打均匀。同时按照每100 μL细胞悬浮液加入5 μg质粒,轻轻吹打均匀备用,电转染参数设置为1 400 V,20 ms,2 pulse。将电转后的细胞接种至预热好的培养皿内,48 h后可在显微镜下观察细胞荧光,分析转染效率。
1.2.5 T7EⅠ实验 T7EⅠ酶 (T7核酸内切酶Ⅰ)可以用来检测基因编辑工具介导的基因突变效率。其检测原理为:先扩增出包含打靶位点的基因序列,T7EⅠ酶可以识别扩增产物中包含的杂交异源双链DNA,并在识别位点进行切割,使酶切产物中包含扩增主带以及酶切后断裂而成的两条较小的DNA条带,通过聚丙烯酰胺凝胶电泳分离出3条大小不一的条带,从而估算出CRISPR/Cas9的打靶效率。
首先设计T7EⅠ引物,使PCR扩增打靶位点共约500 bp的序列长度,使位点位于序列约100~200 bp的位置(表2)。收集细胞并提取基因组,PCR扩增出包含3种基因工具识别位点的Rosa26基因片段,后用胶回试剂盒纯化PCR产物。纯化后的PCR产物高温变性后,经逐步降温退火形成异源双链DNA,在变性退火产物中加入0.5 μL T7EⅠ酶,置于37 ℃水浴锅内,反应30 min。配制w=10%的PAGE胶用于酶切电泳,电泳参数设置为电压120 V,时间90 min。电泳结束后,小心取出PAGE胶,于配制好的核酸染料中染色15 min左右。染色后用清水清洗3次去除残余的核酸染料,置于凝胶成像系统观察并拍照。通过Image J软件估算3种基因编辑工具的切割效率。
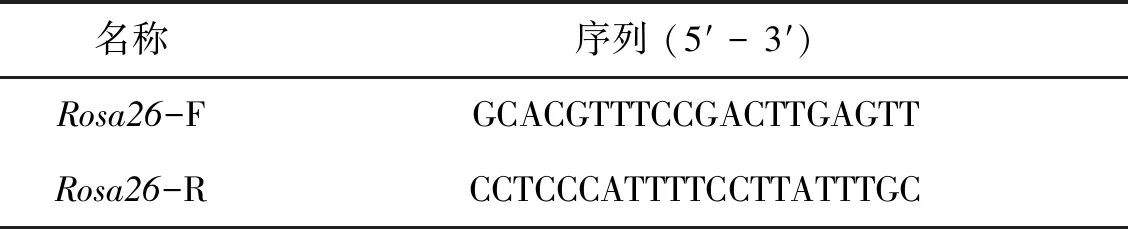
表2 Rosa26编辑位点PCR扩增引物
1.2.6 流式分选 电转后48 h细胞荧光强度较强,可进行流式分选。用胰酶消化贴壁细胞,1 600 r/min离心4 min,吸掉上清,用PBS重悬。将细胞悬液经50 μm尼龙膜过滤到流式管中,以防堵塞流式分选仪,同时准备接收阳性细胞的培养皿,在FACScalibur流式细胞分析仪上可分析细胞的荧光比例与强度。对于要分选培养单克隆的细胞,流式细胞仪设置为96孔板,每孔1个细胞,用添加了含有w=20%胎牛血清的完全培养基的96孔板收集,培养7 d后换液并进一步扩大培养。
1.2.7 嵌合体小鼠制备 促排四周龄母小鼠,并与公鼠合笼,检查交配栓。3.5 d后用引颈法处死母鼠,并收集输卵管和子宫中的胚胎,转移到覆有石蜡油的M16培养基内,37 ℃培养基内培养。
将分选并扩培后的ES单克隆细胞用胰酶消化成单细胞悬液,并转移到M2培养基液滴内,同时将培养好的囊胚转移到液滴内。用显微注射系统将ES细胞注射到囊胚腔内,每个囊胚注射10~12个ES细胞。37 ℃培养1~3 h,待囊胚腔恢复后,移入代孕母鼠子宫中。
2 结果与分析
2.1 ZFN、TALEN、CRISPR/Cas9靶向切割效率鉴定
将构建好的ZFN、TALEN和CRISPR/Cas9表达质粒分别电转染至C2C12细胞中,48 h后收集细胞,提取细胞基因组,通过PCR扩增Rosa26靶位点序列,通过T7EⅠ酶切法分析3种基因编辑工具的靶向切割活性,酶切结果显示ZFN和TALEN-A无明显的靶向切割活性,TALEN-B和CRISPR/Cas9有明显的切割活性,其中CRISPR/Cas9的切割活性略强(图1: B)。进一步的TA克隆测序分析结果验证了T7EⅠ酶切结果,其中ZFN在靶位点引起Indel的效率为4.1%(2/49),TALEN-A为4.2%(2/48),TALEN-B为24%(12/50),而CRISPR/Cas9为30%(12/40)(图1: C)。因此,CRISPR/Cas9在该位点具有相对较高的靶向切割活性。
2.2 ZFN、TALEN、CRISPR/Cas9定点整合外源基因的效率比较
将ZFN、TALEN和CRISPR/Cas9表达质粒分别与同源重组质粒供体共转染至C2C12细胞中,24 h后通过荧光显微镜可观察到少数表达红色荧光蛋白DsRed的细胞(图2:B),表明部分供体质粒在基因编辑工具的介导下通过同源重组正确整合到Rosa26位点。进一步应用流式细胞仪对DsRed阳性细胞的比例进行定量分析,结果显示在C2C12细胞中,ZFN的Knockin效率为(0.11 ± 0.05)%,TALEN-A和TALEN-B的效率分别为(0.13 ± 0.06) %和(0.08 ± 0.02) %,CRISPR/Cas9的效率为(1.36 ± 0.40)%(图2:C),表明CRISPR/Cas9介导的定点整合效率明显高于ZFN和TALEN。
2.3 利用CRISPR/ Cas9构建定点整合外源基因的ES细胞株
由于CRISPR/Cas9具有较强的基因定点整合效率,因此本研究选用CRISPR/Cas9作为基因编辑工具来构建Tyr定点整合的ES细胞。将构建好的pX458-Rosa26质粒与质粒供体共转染至ES细胞中,流式分选出表达有GFP的细胞,继续培养一周后,荧光显微镜下可见在饲养层细胞上,ES细胞开始形成岛状的细胞克隆,部分ES细胞克隆同时表达EGFP绿色荧光蛋白和DsRed红色荧光蛋白(图3: A)。应用克隆环消化带有荧光标记的ES细胞克隆,转移到新的培养皿中,继续培养扩增细胞数量,待其数量足够时,取一部分细胞提取基因组并进行PCR扩增分析。在得到的5个单细胞克隆中,有3个克隆成功扩增出了中间的Tyr基因插入序列,其中#3、#4克隆同时成功扩增出了右侧边界片段(图2:A, 图3:B和图3: C)。其中#3 ES克隆右侧边界片段序列图谱表明外源供体质粒3′端完整地整合到了打靶位点,质粒与打靶位点交界处没有发生碱基的缺失或插入等(图3:D)。

图2 ZFN、TALEN、CRISPR/Cas9 定点整合外源基因的效率鉴定
2.4 构建外源基因整合的嵌合体小鼠
挑选正确整合了Tyr-2A-DsRed基因的3# ES单克隆细胞注射入昆明白小鼠的囊胚腔中,并将囊胚移植到代孕昆明白母鼠输卵管内,最终母鼠成功诞下一只健康存活的嵌合体小鼠,毛色与普通昆明白小鼠明显不同:白毛夹杂着黑毛(图4: A)。近距离观察眼球,其虹膜颜色比对照小鼠明显偏暗(图4:B),表明整合进Rosa26基因的Tyr基因可以正确表达,促进黑色素合成,使嵌合体小鼠表现出不同的毛色表型和虹膜颜色。
3 讨 论
本研究设计了靶向小鼠在Rosa26内含子的ZFN、TALEN、CRISPR/Cas9基因编辑工具,使三者在同一打靶位点对外源Tyr基因进行定点整合。在验证三种基因编辑工具的靶向切割活性时发现设计的ZFN切割活性最低,仅为4.1%;两对TALEN中有一对的切割活性可达到24%;而CRISPR/Cas9的切割活性最强,可达到30%。因此,针对本研究中既定打靶位点设计基因编辑工具,要获得高活性的ZFN难度较大,TALEN居中,CRISPR/Cas9较小。
研究表明,由ZFN或CRISPR/Cas9通过HDR介导的DNA片段的定点整合通常效率较低,尤其在哺乳动物细胞中,通过同源重组实现DNA片段定点整合效率约为10-6~10-5[12-13]。基因编辑工具的选择在一定程度上可以影响定点整合的效率,在同源序列存在的情况下,由基因编辑工具所造成的DNA发生双链断裂能促进同源重组修复,从而提高定点整合外源基因的效率[14]。本研究对红色荧光的分析结果显示,在C2C12细胞中,ZFN和TALEN的介导的定点整合效率约为0.1%~0.3%,CRISPR/Cas9的效率达到2.74%,具有明显的优势,这与前期CRISPR/Cas9具有最佳的切割效率的结果是一致的。
图3 利用CRISPR/ Cas9构建外源基因定点整合ES细胞克隆
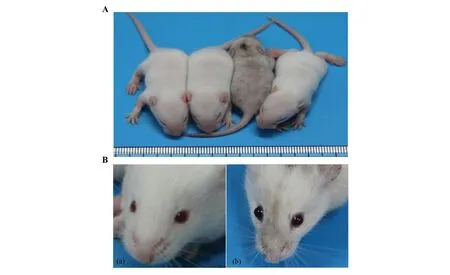
图4 构建Tyr基因定点整合的嵌合体小鼠
使用CRISPR/Cas9对无启动子的质粒供体pTyr-2A-DsRed进行定点整合时,本研究利用pX458载体上携带的EGFP报告基因,通过流式分选可快速富集转染阳性细胞用于后续实验,进一步通过流式分析定点整合到基因组上的供体上的红色荧光蛋白DsRed,即可快速鉴定定点整合的效率。相对于传统的正负药物筛选定点整合的细胞单克隆,此方法省去繁琐的筛选过程,更加省时高效。
对于定点整合的细胞基因组的巢式PCR扩增得到了3′端边界序列和中间插入序列的条带,证明了外源基因的成功整合。而针对5′端边界序列的扩增无明显目的条带,我们推测可能是由非常规重组(illegitimate recombination)引起的。非常规重组与同源重组在哺乳动物细胞DSB修复过程中互为竞争关系,研究表明,非常规重组可能导致在基因组与donor连接位置处发生复杂的DNA重组,包括DNA缺失,重复,插入和倒位等[15-16]。当然左侧同源臂的大量GC重复序列也在一定程度上增加了扩增的难度。
最后,我们成功得到了一只整合了Tyr基因的嵌合体小鼠,该小鼠相对于非嵌合体小鼠具有明显的毛色区别。通过观察毛色变化可以更加直观、快速地鉴别目标外源基因整合小鼠,这为今后制作Rosa26定点整合小鼠提供了一种简便高效的方法。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
基层中医药(2022年4期)2022-07-22
舰船科学技术(2022年11期)2022-07-15
中国土壤与肥料(2021年5期)2021-12-02
农业资源与环境学报(2021年5期)2021-10-06
汉字汉语研究(2021年2期)2021-08-30
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
汉字汉语研究(2019年2期)2019-08-27
