
骨髓增殖性肿瘤的分子诊断进展
2020-06-28 12:41钱军
诊断学(理论与实践) 2020年2期
钱 军
(江苏大学附属人民医院血液科,江苏 镇江 212002)
骨髓增殖性肿瘤 (myeloproliferative neoplasm,MPN)是一组表现为粒系、红系和(或)巨核系过度增生的克隆性造血干细胞疾病。世界卫生组织(World Health Organization,WHO)造血与淋巴组织肿瘤相关分类及诊断(以下简称WHO 相关分类)2001 版将此类疾病定义为慢性骨髓增生性疾病;WHO 相关分类2008 版则将其确定为MPN。最新的WHO 相关分类2016 版分型中MPN 包括7个亚型[1],即慢性髓细胞性白血病(chronic myelogenous leukemia,CML)、慢性中性粒细胞性白血病(chronic neutrophilic leukemia,CNL)、真性红细胞增多症(polycythemia vera,PV)、原发性血小板增多症(essential thrombocythemia,ET)、原发性骨髓纤维化(primary myelofibrosis,PMF)、慢性嗜酸性粒细胞白血病-非特指型 (chronic eosinophilic leukemia,not otherwise specified,CEL-NOS)、骨髓增殖性肿瘤不能分类型(myeloproliferative neoplasms,unclassifiable,MPN-U)(见表1)。
MPN 发病是由于造血干细胞(hematopoietic stem cell,HSC)驱动基因异常所产生的一系或多系髓系成熟细胞克隆性扩增,而不同的驱动基因重排或突变导致的疾病临床特征、并发症和疾病进展的风险各不相同,但在疾病初始阶段和发展过程中,不同亚型MPN 患者的临床表现经常重叠、影响疾病诊断。
CML 的分子诊断
一、t(9;22)染色体易位
90%~95%的CML 患者具有特征性的t(9;22)染色体易位[即费城染色体(Philadelphia chromosome,Ph 染色体)],其余易位则为变异型或隐匿型,因而采用荧光原位杂交或反转录聚合酶链反应可能检出BCR-ABL1 融合基因。绝大多数的CML 可见BCR 基因外显子12~16(M-BCR)断裂所形成的融合基因蛋白p210,罕见病例(<2%)可见BCR 外显子1~2 断裂(约1.2%)形成的融合基因蛋白p190、外显子17~20 断裂形成的p230 融合蛋白(约0.3%)、外显子6 断裂形成的p185 蛋白,亦有报道e8a2 等其他融合基因等[2-4]。因此,当常规核型分析未能检出典型或变异型t(9;22),荧光原位杂交未能发现BCR-ABL1融合信号,且反转录聚合酶链反应未能检出p210、p190、p230 融合蛋白转录本时,则排除CML 的可能性大。

表1 MPN 的历年WHO 分型
二、BCR-ABL1 重排和JAK2 突变
BCR-ABL1 重排和JAK2 突变分别是Ph 染色体阳性(+)CML 和Ph 染色体阴性(-)MPN 发病的不同驱动因素,一般认为两者不会共存。2018 年,美国对2005 年至2015 年间1 570例疑诊为MPN 的患者开展多中心的研究揭示,BCR-ABL1 和JAK2 共阳性率达0.4%[5]。2017 年,德国一项研究在10 875例确诊的MPN 患者中发现,有0.2%的患者为BCRABL1 和JAK2 突变共阳性[6]。但在此之前,意大利的研究者在BCR-ABL1 阳性CML 患者中发现JAK2 V617F 突变率达2.5%(8/314例)[7]。由于CML患者一般不会同时检测JAK2 突变,因此目前研究者对于BCR-ABL1 重排和JAK2 突变共存的确切发生率并不明确。迄今为止,已有超过40例BCRABL1 合并JAK2 突变的MPN 病例被报道,43%的患者为同时存在,39%的患者为先有JAK2 突变(多为PV)后继发BCR-ABL1 重排,18%的患者为先有BCR-ABL1 重排后继发JAK2 突变(多为骨髓纤维化和ET)[5,8]。因此,当CML 患者经酪氨酸激酶抑制剂治疗后无明显改善,或者改善后再次出现白细胞和(或)血小板计数增高、脾脏肿大,除了怀疑疾病进展外,也要排除继发或转为其他MPN 的可能。
CNL 的分子诊断
CNL 是一种罕见的MPN,其特征是外周血中性粒细胞持续性增多以及由中性粒细胞所致的骨髓过度增生和肝脾肿大[9]。诊断CNL 时,需要排除反应性中性粒细胞增生、BCR-ABL1p230相关的中性粒细胞性慢性髓细胞性白血病 (neutrophilic CML,CML-N)、骨髓增生异常综合 征(myelodysplastic syndrome,MDS)/MPN(见表2)。
现在已知CNL 的发病与CSF3R 突变明确相关,而该基因突变存在膜近端点突变和截断型突变2 种类型。前者多位于第14 外显子,T618I 和T615A常见,这些突变导致受体发生非配体依赖性的自发性活化,激活Janus 激酶/信号转导及转录激活(Janus kinase/signal transducers and activators of transcription,JAK-STAT)途径、诱导增殖和生存信号,携带此突变的患者对芦可替尼治疗敏感;后者多位于第17 外显子,主要为无义突变或移码突变(如D771fs、S783fs、Y752X、W791Z),导致转录提前终止,从而形成截断型CSF3R 胞内段,而截断型CSF3R 发生过表达,在与粒细胞集落刺激因子(granulocyte colony stimulating factor,G-CSF) 结合后激活SFK-TNK2 途径,携带此类型突变的患者对达沙替尼治疗敏感[9-10]。88%的CNL 患者为CSF3R第14 外显子T618I 突变,其中大多数患者为单独发生,少数可合并截断型突变;部分CNL 患者可合并 SETBP1、ASXL1、SRSF2、TET2、U2AF1、NRAS 和CBL 突变[9-10]。尽管CSF3R 突变亦可发生于不典型慢性髓细胞性白血病 (atypical chronic myelogenous leukemia,aCML)患者,但鉴于CSF3R T618I 等活化型突变在CNL 中的特异性,WHO 2016 版相关诊断标准将其确定为CNL 的诊断性分子标志物。因此,如果在aCML 患者中发现CSF3R 突变,则要对其进行仔细的形态学评价,以防将CNL 误诊为aCML。
CEL-NOS 的分子诊断
一系列反应性或克隆性疾病可发生嗜酸性粒细胞增多症(hypereosinophilia,HE),引起HE 的疾病众多。嗜酸性粒细胞浸润和颗粒物释放可能导致危及生命的器官损伤[11]。2011 年,嗜酸性粒细胞疾病和综合征工作组会议将除了嗜酸性粒细胞增多综合征(hypereosinophilic syndrome,HES) 之外的HE 分为4 种类型,即遗传性(家族性)HE(HEFA)、意义不明的HE(HEUS)、原发性[克隆性和(或)肿瘤性]HE(HEN)、继发性(反应性)HE(HER)[12]。这4 种类型并不代表最终诊断,而是为了指导进一步的诊断评估。CELNOS 是排他性诊断,必须排除其他伴有HE 的髓系肿瘤或淋系肿瘤(myeloid or lymphoid neoplasms associated with eosinophilia,MLN-eo)、造血干细胞肿瘤,如伴有HE 和基因重排的MLN-eo (包括PDGFRA、PDGFRB、FGFR1 重排 或PCM1-JAK2 的MLN)、CML-eo、AML-M4-eo、伴有HE 的系统性肥大细胞增多症、MPN-eo。PDGFRA、PDGFRB、FGFR1重排或 PCM1-JAK2、BCR-ABL1、CBFβ MYH11、c-KIT D816V 突变、JAK2 V617F 突变等可资鉴别。
当前,研究者对CEL-NOS 的基因异常尚缺乏系统性研究,在嗜酸性粒细胞增多的特定背景下,ASXL1、TET2、EZH2等基因突变的检出有助于CEL-NOS 的诊断。但必须注意DNMT3A、ASXL1、TET2 突变是非血液肿瘤老年人群中意义未明的克隆性造血(clonal hematopoiesis of indeterminate potential,CHIP)的常见突变,如检出,要仔细排除合并HER。最近,欧洲学者在1 715例HE 患者中发现,27例 (7例初始诊断为HES、20例为髓系肿瘤)患者可检出STAT5B N642H 突变,并且对4例患者的T 细胞进行了STAT5B N642H 突变检测,结果提示4例均为阴性,这些患者的总体生存时间仅为30个月,故建议将伴STAT5B N642H 突变的伴HE 髓系肿瘤归类为CEL-NOS[13]。

表2 CNL、aCML、CMML 和MDS/MPN-U 比较
PV 的分子诊断
一、JAK2 突变
红细胞增多通常反映了体内红细胞总量的增加(红细胞增多症),但有时可能是由于血浆体积减少(假性红细胞增多症)。红细胞增多症由多种遗传性和后天性疾病引起,可分为原发性或继发性。检测内源性促红细胞生成素 (erythropoietin,EPO)浓度,如降低,则倾向排除继发性因素。超过95%的PV患者存在JAK2 V617F 突变,3%的患者具有JAK2外显子12 突变,JAK2 V617F 突变或外显子12 突变的检出有助于PV 诊断[14]。最近一项包含27 078例病例的荟萃分析揭示,PV 中的JAK2 V617F 突变检出率在46.7%~100%[15],而该检出率差异较大可能由于以下3 点原因。①方法学不同;②存在JAK2外显子12 突变或其他罕见位点突变;③未能排除其他先天性疾病。因此,如患者的EPO 浓度降低而JAK2 V617F 和外显子12 突变检测为阴性时,需排除先天性促红细胞生成素受体突变;如EPO 浓度正常而JAK2 V617F 和外显子12 突变检测为阴性,则要进一步排除氧调节相关基因VHL 的先天性突变以及高氧亲和血红蛋白病、高铁血红蛋白血症、2,3-双磷酸甘油酸变位酶缺乏症。
二、其他分子
除了JAK2 突变之外,PV 患者不存在MPL 突变和CALR 突变,但可发生DNMT3A、TET2、ASXL1、SH2B3、CEBPA、SRSF2、IDH2、CBL 等突变[16-18],其 中ASXL1、SRSF2 和IDH2 突变可能与骨髓纤维化转化及白血病进展相关[17-18]。
ET 的分子诊断
ET 是巨核系优势增生伴血小板计数增高(≥450×109/L)的MPN,鉴别诊断需排除引起血小板增多的其他克隆性或反应性原因,尤其是需要与纤维化前期PMF(pre-PMF)及隐性PV(masked PV)鉴别,主要是通过骨髓组织病理学检查进行区分,基因异常检出无助于ET 与pre-PMF、PV 的鉴别。绝大多数ET 是由JAK2、CALR 或MPL 的体细胞突变所驱动,JAK2 V617F、CALR 和MPL W515 突变发生率分别为50%~60%、25%~35%和3%左右,其检出有助于ET 的诊断。10%~12%的ET 患者为三基因(JAK2、CALR 和MPL)突变阴性,但需要注意的是,许多单位的检测是针对三基因的热点突变位点(JAK2 V617F 和外显子12 突变、MPL W515L/K/R/A/N/G/S 突变、CALR 基因52 bp 缺失的Ⅰ型突变L367fs*46 和5bp 插入的Ⅱ型突变K385fs*47)。因此,即便是在所谓的“三阴性”患者中,如果针对全部外显子均进行测序的话,仍可能检出其他位点的罕见突变,如JAK2 G127、CALR S189N/E381A、MPL T119/S204/E230/Y252/E259/S505/Y591 等[19-21],这些少见位点突变的检出也有助于确定疾病的克隆性,排除反应性血小板增多。
此外,研究者对于JAK2、MPL 和CALR 胚系突变的影响认识还不足,胚系突变的检出要求临床医师需小心求证遗传性或家族性血小板增多症可能,如家族性ET 相关的JAK2 T875N 胚系突变可引起STAT3、STAT5 活性增强并促进细胞增殖[22]。JAK2、CALR、MPL 三基因突变可伴随 TET2、ASXL1、DNMT3A、SF3B1、CEBPA、SH2B3、ZRSR2、CSF3R、EZH2、TP53、IDH2、U2AF1 等基因突变,但如JAK2、CALR、MPL 中任意基因伴随SF3B1 突变时需谨慎鉴别ET 与MDS/MPN-环形铁粒幼细胞(ringed sideroblasts,RS)-血小板增多(thrombocytosis,T)。ET 患者检出SH2B3、IDH2、SF3B1、U2AF1、EZH2 和TP53 突变提示可能预后不良[17]。此外,天津血液研究所报道,JAK2、CALR、MPL 三基因突变阴性患者中常见 TET2 (33.82% )、SH2B3(29.41%)和ASXL1(23.53%)突变[21]。
PMF 的分子诊断
骨髓纤维化分为原发性和继发性,后者可由感染、自身免疫性疾病、毛细胞白血病或其他淋巴肿瘤、转移性肿瘤等所致。WHO 相关分类2016 版将PMF 分为pre-PMF 和明显的PMF(overt-PMF)。除要排除继发性骨髓纤维化外,PMF 尤其是pre-PMF还要与ET、MDS/MPN-RS-T、慢性粒单核细胞白血病(chronic myelomonocytic leukemia,CMML)等 相鉴别(见表3)。目前只能通过单核细胞计数、巨核细胞形态、红系增生及发育异常等形态学特征来进行区分,这些疾病在分子改变上具有较大重叠。JAK2 V617F、CALR 和MPL W515 基因突变发生率在PMF 中分别为50%~60%、30%左右和5%~8%[1,23-24]。约12%的PMF 患者为JAK2、CALR、MPL三阴性,可有ASXL1(31%)、SRSF2(17%~23%)、SF3B1(7%~14%)、U2AF1(5%~16%)、EZH2(5%~7%)突变[1,25-26]。CALR 突变阴性和ASXL1、SRSF2、EZH2、IDH1/IDH2 突变阳性已作为预后不良因素被纳入MDS 的国际预后指数系统 (MDS international prognostic scoring system,MIPSS)70+V2.0预后系统;CALR 突变阴性以 及ASXL1、SRSF2、U2AF1Q157 突变阳性作为不良因素被纳入遗传预测评分系统(genetically inspired prognostic scoring system,GIPSS)[26]。
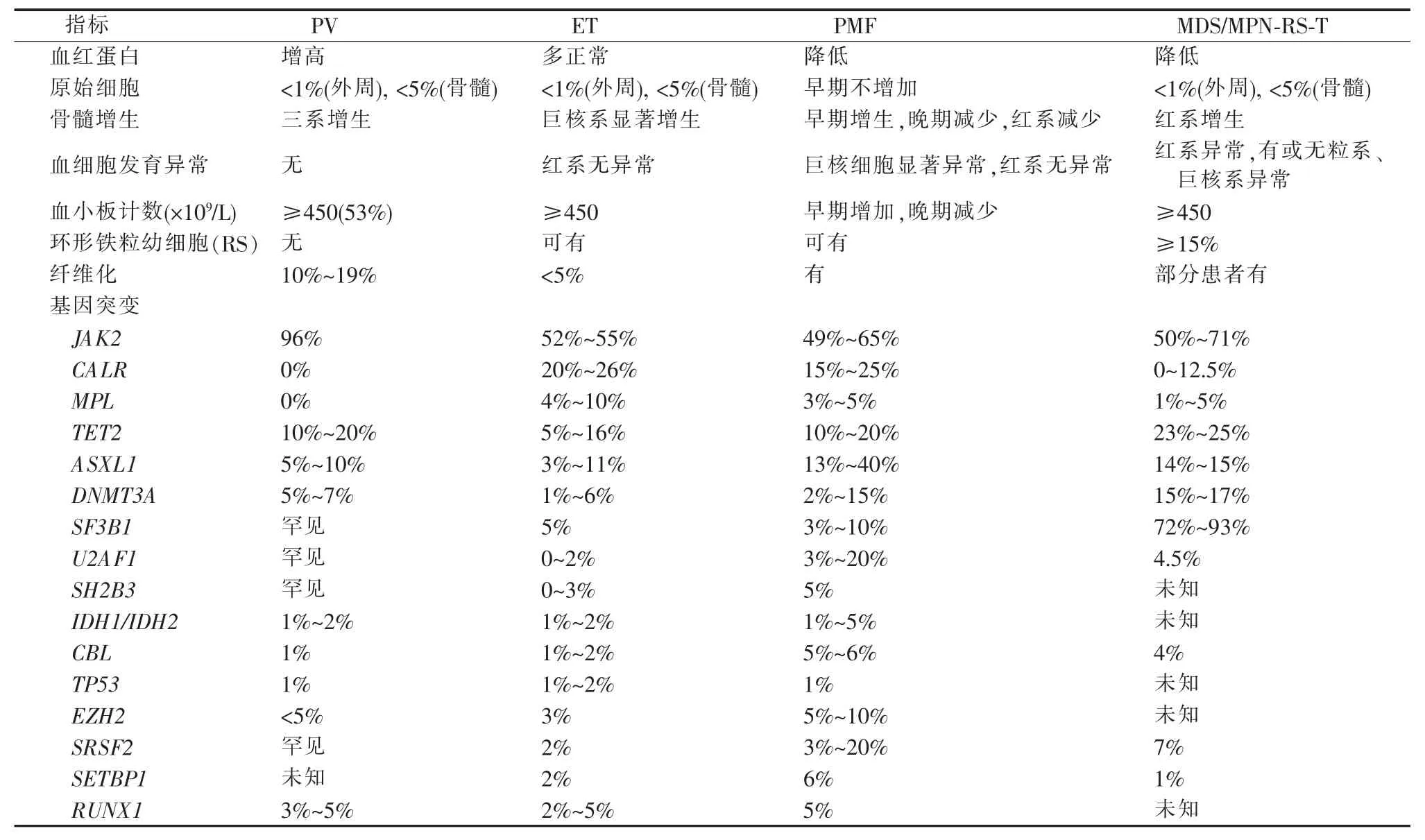
表3 PV、ET、PMF 和MDS/MPN-RS-T 比较
MPN-U 的分子诊断
WHO 相关分类2016 版将那些具有明确的MPN 临床、实验室、形态和分子特征但不符合任一特定MPN 亚型诊断标准的病例,或者具有2个或多个MPN 亚型重叠特征的病例归入MPN-U[1]。既往认为,MPN-U 占MPN 的8.6%~30.0%[27-28],但WHO相关分类2016 版的精确分型使MPN-U 比例减少[28]。目前,MPN-U 通常包括3个亚类:①PV、prePMF或ET 的早期阶段,其临床表现尚不符合各自的诊断标准;②MPN 晚期阶段,由于显著的骨髓纤维化、骨硬化或原始细胞增多等疾病进展和(或)出现发育异常使潜在的疾病被掩盖;③具有MPN 的明确证据,但由于并存的肿瘤或炎症性疾病使常见的诊断性临床和(或)形态学特征被掩盖。如果患者不符合某一特定亚型的MPN 诊断标准,则必须首先考虑其很可能不是MPN,要排除感染、炎症、药物、肿瘤等所致的骨髓反应性增生。JAK2、CALR或MPL 突变可确立MPN-U 诊断,其发生率分别为67%~72%、9%~11%和0~3%;在三基因突变阴性的情况下,髓系肿瘤相关的其他突变检出 (如ASXL1、EZH2、TET2、IDH1/IDH2、SRSF2、SF3B1 等)有助于确认克隆性造血的存在,但目前缺乏MPNU 基因突变谱的系统性研究。
此外,获得性克隆性突变也可能发生于没有髓系肿瘤的健康老年人的造血细胞中。因此,对于基因突变分析要在特定的临床背景下谨慎解读。
结语
MPN 是由不同驱动基因突变或重排引起的一组异质性克隆性疾病,在生物学及疾病表型上复杂多样。随着遗传学技术的不断进步,MPN 的概念和分类得以不断更新,病理生理学认识得以不断深入,诊断、预后评估和治疗管理得以不断完善。
尽管在MPN 生物学方面取得了极大进展,但一些重要的基本问题如相同的遗传事件 (如JAK2 V617F 突变)如何导致不同的临床表型仍然未得到很好地解决。单细胞测序对于阐明MPN 中不同分化细胞的克隆构成、克隆演变将会产生巨大的推动作用。对于这些问题的解答也必将会产生临床转化,使MPN 患者的疾病管理、并发症处理、预防及精准个体化治疗研发得到进一步改进。
猜你喜欢
火控雷达技术(2022年2期)2022-07-22
中国生殖健康(2020年4期)2021-01-18
小学生导刊(2018年13期)2018-06-29
电脑知识与技术·经验技巧(2018年3期)2018-06-06
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
湖北农业科学(2014年11期)2014-09-10
消费导刊(2009年1期)2009-03-06
