
提高酿酒酵母异戊二烯产量的代谢途径的挖掘
2020-07-06 13:25曹小贺张海波包文智刘丽娟夏海锋
食品与发酵工业 2020年12期
曹小贺,张海波,包文智,刘丽娟*,夏海锋*
1(工业生物技术教育部重点实验室(江南大学),江苏 无锡,214122)2(江南大学 生物工程学院,江苏 无锡,214122) 3(生物基材料重点实验室(中国科学院青岛生物能源与过程研究所),山东 青岛,266101) 4(内蒙古师范大学 生命科学与技术学院,内蒙古 呼和浩特,010022)
异戊二烯是一种具有挥发性和疏水性的萜类烃分子[1]。异戊二烯在橡胶生产、香水和农药制造等领域有着广泛的工业应用[2]。目前异戊二烯的生产方法主要有C5馏分分离法、脱氢法和化学合成法[3-5]。工业合成异戊二烯主要是来自石油裂解C5组分[6]。但随着石油资源的日益枯竭及其使用所带来的污染使得异戊二烯的合成受到限制。近年来随着合成生物学及代谢工程的发展,生物法合成异戊二烯作为一种绿色可持续的异戊二烯生产方法已经成为研究热点。
目前生物法合成类异戊二烯化合物的宿主菌主要是酿酒酵母和大肠杆菌。其中酿酒酵母作为真核类模式菌株,不仅具有遗传背景清晰和高糖代谢分解速率等优点[7],而且它具有天然的甲羟戊酸(mevalonate,MVA)途径,是生产异戊二烯类产品的良好宿主。乙酰辅酶A(Acetyl-CoA)作为甲羟戊酸途径的第一个前体物质,是合成异戊二烯类化合物所必需的。在酿酒酵母中Acetyl-CoA 分布在细胞质、线粒体、过氧化酶体及细胞核4个不同的亚细胞区域[8]。在线粒体中,Acetyl-CoA 是由丙酮酸脱氢酶复合体(pyruvate dehydrogenase,PDH)催化丙酮酸生成的。线粒体具有Acetyl-CoA含量丰富,能阻止中间产物进入竞争途径等优点。有不少学者通过充分利用线粒体Acetyl-CoA来提高目标产物的产量。LV等[1]通过在酿酒酵母线粒体中引入MVA途径,显著提高了异戊二烯的产量。AVALOS等[9]在酿酒酵母中将Ehrlich途径引入到线粒体中,使异丁醇产量得到了提高。虽然在线粒体中合成目标产物有一定益处,但往往需要将代谢途径引入线粒体中,操作复杂,同时存在过多中间体的积累会抑制细胞生长的问题[10]。而在酵母细胞质中合成目标产物对细胞的生长没有影响。在酵母细胞质中,Acetyl-CoA是由乙酰辅酶A合成酶(Acetyl-CoA synthetase,ACS)合成的[7]。LV等[11]通过过表达基因ADH2、ALD6、ACS1和ACS2,显著提高了酿酒酵母中异戊二烯的产量;SHIBA等[12]通过在酿酒酵母中过表达乙醛脱氢酶和来自肠道沙门氏菌的Acetyl-CoA合成酶,使得紫穗槐二烯产量增加了1.2倍。MEADOWS等[13]过表达木酮糖-5-磷酸特异性磷酸转酮酶基因(5-phosphate-speific-phosphoketolase)(LmPK)和磷酸转乙酰酶(phosphotransacetylase)(CkPTA)基因,从5-磷酸-木酮糖经LmPK和CkPTA催化合成Acetyl-CoA并将碳流重新定向,提高了酿酒酵母中法尼烯的产量。此外,CARDENAS等[14]通过敲除酿酒酵母中一系列与丙酮酸及Acetyl-CoA线粒体转运相关的基因增加细胞质中Acetyl-CoA的通量,提高了聚酮类化合物三乙酸内酯的产量,其中MPC2、POR2、PDA1和YAT2基因具有最好的效果。
综上,本研究以来源于银白杨的ispS为基础,通过敲除线粒体丙酮酸载体(MPC2)、线粒体孔蛋白 (POR2)和丙酮酸脱氢酶复合体的E1α亚基(PDA1)基因来切断Acetyl-CoA往线粒体的支流,增加细胞质中Acetyl-CoA的通量。根据摇瓶发酵结果,进而在细胞质中引入来自Leuconostocmesenteroide和Clostridiumkluyveri的木酮糖5-磷酸特异性磷酸转酮酶基因LmPK和磷酸转乙酰酶基因CkPTA合成Acetyl-CoA来进一步增加通量。然后在此基础上结合大肠杆菌IDI1进行发酵验证。本研究结合代谢工程,旨在探索提高酿酒酵母异戊二烯产量的代谢途径,为酿酒酵母合成异戊二烯工业化奠定一定的基础。
1 材料与方法
1.1 材料与试剂
1.1.1 主要试剂
异戊二烯,Aladdin公司;YNB 培养基,索莱宝有限公司;Drop-out-His /URA(Do-His/URA),Clotech公司;限制性内切酶、T4DNA连接酶,宝生物工程(大连)有限公司;无缝克隆试剂盒、高保真DNA聚合酶、质粒提取试剂盒、胶回收试剂盒,南京诺唯赞生物科技有限公司。
1.1.2 菌株与质粒
菌株与质粒如表1和表2所示。

表1 本研究中所用的菌株Table 1 Strains that used in this study

表2 本研究中所用的质粒Table 2 Plasmids that used in this study
1.1.3 培养基
Luria-Bertani(LB)培养基:酵母粉 5 g/L,蛋白胨 10 g/L,NaCl 10 g/L,需要时加入抗生素(氨苄青霉素 100 μg/mL 卡那霉素 50 μg/mL)。筛选培养基:YNB 6.7 g/L[含(NH4)2SO4],葡萄糖 2 g/L,115 ℃灭菌 20 min,DO-His/URA 0.065 g/L,氨基酸溶液过滤除菌,固体培养基均在液体培养基中添加 2%(质量分数)的琼脂粉;发酵培养基同筛选培养基,但把葡萄糖换为半乳糖。酵母浸出粉胨葡萄糖(yeast peptone dextrose,YPD)液体培养基:蛋白胨 20 g/L,酵母粉 10 g/L,葡萄糖 20 g/L,115 ℃ 灭菌 30 min,固体培养基均在液体培养基中添加 2%(质量分数)的琼脂粉。
1.1.4 仪器与设备
PCR 基因扩增仪,美国 Bio-Rad 公司;气相色谱仪,美国安捷伦;紫外可见分光光度计,美国 Varian公司;ALLegra X-22 型台式冷冻离心机,美国 Beckman 公司;凝胶成像系统,美国 Bio-Rad 公司;LDZM-80KCS 型蒸汽灭菌器,上海申安医疗器械厂;WD-9405B 型水平摇床,北京六一仪器厂。
1.1.5 摇瓶培养条件
种子液的制备:将重组菌从 DO-His 筛选平板上挑取单菌落至 5 mL 筛选液体培养基中,30 ℃、200 r/min 培养 24 h 后,即为种子液。重组菌发酵实验:将种子液4 000 r/min离心5 min, 用无菌ddH2O重悬2次,然后转接到 50 mL发酵培养基中,使初始 OD600为 0.1,在 30 ℃、200 r/min 培养 72 h。
1.2 实验方法
1.2.1 目的基因扩增与重组
将来自Populusalba的ispS序列[15]进行合成并将密码子按照酿酒酵母偏好性进行优化,以ispS-F,ispS-R扩增ispS基因序列,获得基因长度为1 788 bp的ispS基因。PCR 程序:95 ℃预变性 5 min;95 ℃变性30 s;55 ℃退火 30 s,72 ℃延伸2 min,34个循环;72 ℃后延伸 5 min。PCR产物经胶回收纯化后,连接载体pESC-His,小量抽提质粒酶切鉴定,进行 DNA 测序。以大肠杆菌基因组为模板,用EcIDI1-F、EcIDI1-R扩增EcIDI1,获得大小549 bpEcIDI1基因。PCR程序同上,但延伸时间为30 s。PCR 产物经胶回收纯化。以pUG6质粒为模板,用MPC2-F和MPC2-R分别扩增含有MPC2的上游、下游50 bp序列的KanMX基因,同理分别用PDA1-F、PDA1-R和POR2-F、POR2-R扩增KanMX基因。PCR程序同上。PCR 产物经胶回收纯化(引物序列见表3)。

表3 实验所用引物Table 3 Primer used in this study
注:下划线部分为酶切位点
1.2.2 重组菌株构建
酿酒酵母转化均采用醋酸锂化学转化方法[16]。转化后涂布于相对应的氨基酸缺陷 YNB 平板和其相应抗生素平板,挑选转化子,采用碱性水溶液破壁,具体操作为将挑取的单菌落置于 20 μL 30 mmol/L NaOH 溶液中,95 ℃、5 min,立即置于4 ℃、1 min,循环3~5次。
1.2.3 气相色谱检测
色谱条件[17]为Agilent 7890B. DB-5色谱柱(30 m×0.25 mm×0.25 μm);程序升温条件为初始温度50 ℃,保持30 s,以8 ℃/min升温至100 ℃,立即以30 ℃/min升温至200 ℃,保持3 min;进样口温度为150 ℃,载气(氮气)流速为1 mL/min;不分流模式。采用外标法测定异戊二烯样品的浓度,计算结果取 3 个平行发酵实验的平均值。
2 结果与分析
2.1 目的基因的克隆和表达载体构建
目前研究表明银白杨的异戊二烯合酶具有较好的酶活性[18],我们将银白杨的ispS[15]经华大基因合成并优化,并以优化后的ispS为模板,采用 PCR 法扩增ispS基因,扩增出特异性条带,大小为1 788 bp。将PCR 产物经BamH I 和SalI 酶切并纯化后,与经相同酶切的载体 pESC-His 连接。在含有氨苄抗生素的 LB 平板上筛选阳性克隆,并通过菌落 PCR 鉴定阳性克隆(图略)。抽提重组克隆菌株质粒,经BamH I和SalI酶切得到1 788 bp和6 684 bp两个片段(图1-a),说明ispS基因与载体pESC-His相连,经测序验证正确,载体pESC-His-ispS构建成功。以大肠杆菌基因组为模板,用EcIDI1-F、EcIDI1-R扩增EcIDI1大小为549 bp (图略)。将 PCR 产物纯化后,与经SpeI 和SalI 酶切的载体 pESC-His-ispS采用无缝克隆的方式连接。鉴定阳性克隆步骤同上(图略)。抽提重组克隆菌株质粒分别经BamH I 和SacI酶切,得到1 576 bp和7 728 bp的片段(图1-b),大小符合预期,经测序验证正确。验证EcIDI1基因与载体pESC-His-ispS相连,载体pESC-His-ispS-EcIDI1构建成功。

M-DL10000 DNA marker; 1-pESC-His-ispS质粒双酶切; 2-pESC-His-ispS-EcIDI1质粒双酶切 a-BamH I 和SalI 双酶切产物;b-BamH I 和SacI 双酶切产物
图1 质粒酶切验证琼脂糖凝胶电泳图
Fig.1 The agarose gel electrophoresis of plasmids enzyme digestion
2.2 PDA1, MPC2, POR2基因的敲除
以KanMX作为筛选标记进行敲除基因是个传统方法,其无需敲除或突变酵母内源基因,不损伤菌株遗传背景,是可替代营养缺陷型的理想遗传标记[19-20],通常选择以目的基因上下游500 bp作为同源片段,而本研究中,我们选择上下游50 bp的同源片段进行敲除。如图2所示,以pUG6质粒[21]为模板,用MPC2-F和MPC2-R扩增KanMX基因,约1 700 bp。同理,用PDA1-F、PDA1-R和POR2-F、POR2-R扩增,获得相应的KanMX片段。然后用醋酸锂化学转化方法将PCR扩增出来的3个片段分别转入CEN.PK2-1C中,30 ℃培养2~4 d。然后挑取转化子,酿酒酵母 CEN.PK2-1C 基因组作为对照,以MPC2-TEST-F、MPC2-TEST-R进行菌落PCR验证,对照组扩增出目的片段,大小为232 bp,而敲除菌株未扩增出目的条带,说明MPC2基因敲除成功。同理,用PDA1-TEST-F、PDA1-TEST-R和POR2-TEST-F、POR2-TEST-R进行验证,对照组扩增出大小分别为470 bp和 328 bp的条带,而敲除菌株未扩增出目的条带,说明敲除PDA1、POR2基因成功。这也证明了我们敲除设计的正确性,选取50 bp的同源片段也能准确敲除目的基因,且设计更为便捷。
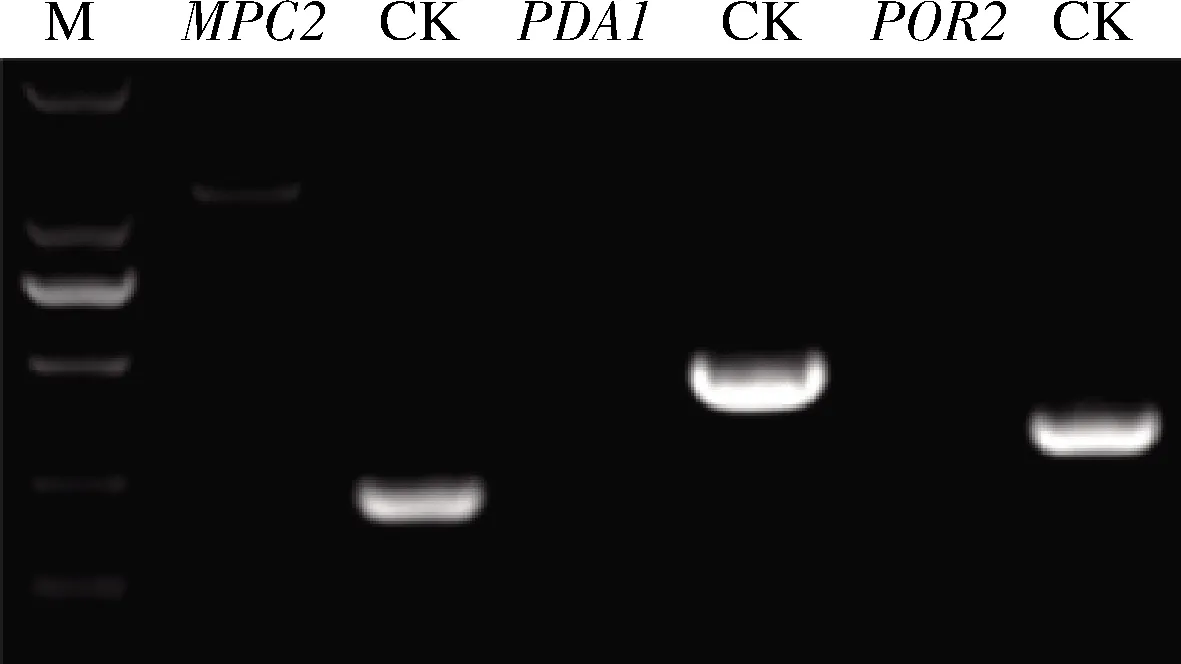
M-DL2000 DNA marker; CK-对照
图2MPC2、PDA1和POR2基因的敲除验证 琼脂糖凝胶电泳图
Fig.2 The agarose gel electrophoresis of knockout verification ofMPC2,PDA1andPOR2genes
2.3 GAL80的敲除及LmPK和CkPTA的过表达
CRISP-Cas9是一种近年来兴起的基因编辑技术,具有使用方便、构建简单、可以覆盖大多数区域的基因编辑需求和成本低等优点[22],因此我们选用该方法进行GAL80的敲除工作,该方法以 URA 为筛选标记,可以在敲除基因的基础上过表达其他基因,已经成为酵母中常用的基因敲除并过表达基因的方法[23-24]。将实验室保存的pUC19-ALD6US-TADH1-CkPTA-PGAL1-PGAL10-LmPK-TCYC1-ALD6DS 和 pUC19-GAL80US-TADH1-PGAL1-PGAL10-AaFS-TADH1-GAL80DS 质粒分别以SacI,SacⅡ 进行酶切,对酶切产物 TADH1-CkPTA-PGAL1-PGAL10-LmPK-TCYC1和 pUC19-GAL80US-GAL80DS 胶回收纯化。然后用 T4DNA Ligase连接,获得载体pUC19-GAL80US-TADH1-CkPTA-PGAL1-PGAL10-LmPK-TCYC1-GAL80DS,然后用引物 LC-GAL80-F、LC-GAL80-R 扩增出片段GAL80US-TADH1-CkPTA-PGAL1-PGAL10-LmPK-TCYC1-GAL80DS,采用 CRISPR-Cas9方法[25]敲除GAL80基因,分别将 2 μL pML104 (SwaI、BclI 酶切),10 μLGAL80-gRNA,18 μLGAL80US-TADH1-CkPTA- PGAL1-PGAL10-LmPK-TCYC1-GAL80DS 片段一起转入△PDA1酵母感受态细胞中,涂 DO-URA平板,30 ℃培养 2~4 d。挑取转化子,以 TEST-GAL80-F、TEST-GAL80-R 进行 PCR 验证基因GAL80,对照组扩增出目的片段,大小为 458 bp,而敲除菌株未扩增出目的条带,表明基因GAL80被成功敲除,然后分别以 TEST1-F、TEST1-R 和 TEST2-F、TEST2-R 进行 PCR,验证片段GAL80US-TADH1-CkPTA-PGAL1,10-LmPK-TCYC1-GAL80DS是否被整合到基因GAL80处,验证原理如图3所示。电泳图结果表明,敲除菌株分别扩增出大小为1 321 bp和1 500 bp的条带,而对照组未扩增出目标条带(图4),说明敲除GAL80基因并过表达LmPK和CkPTA基因成功。
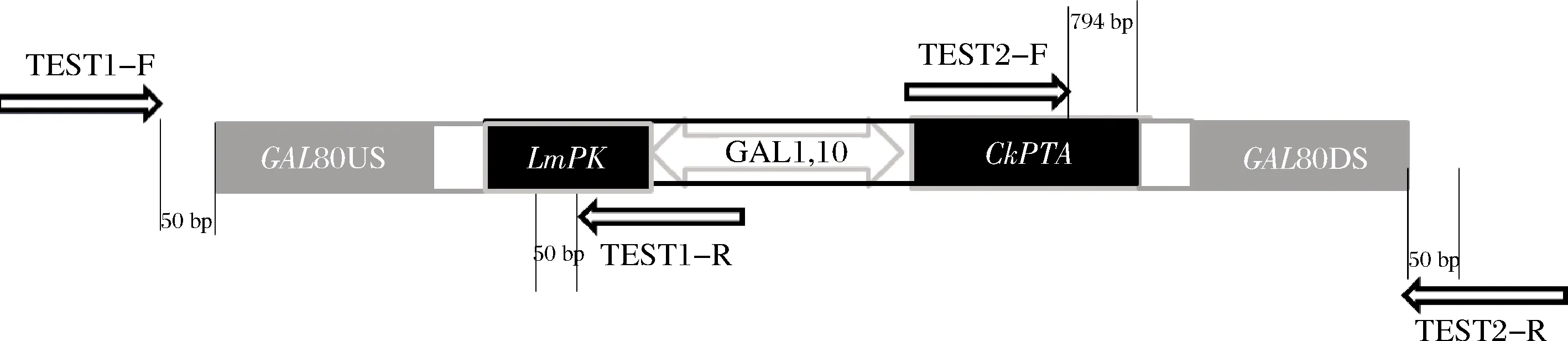
图3GAL80 基因的敲除验证原理图
Fig.3 Schematic diagram ofGAL80 gene knockout verification
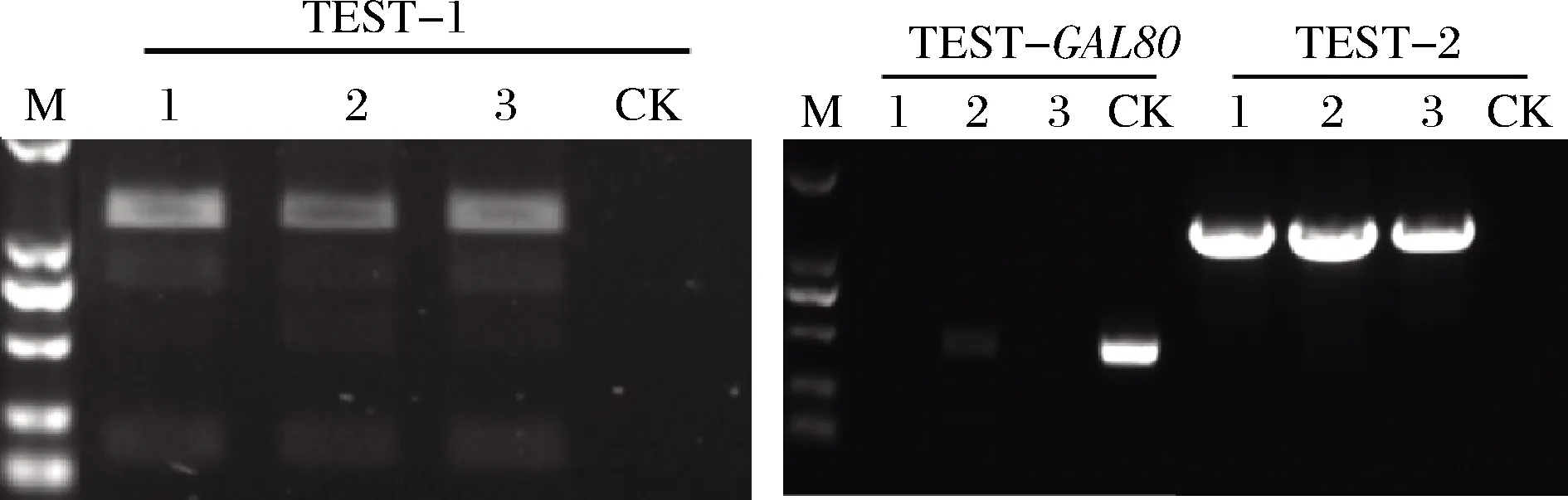
M-DL2000 DNA marker; CK-对照;1~3-菌株编号
图4GAL80基因的敲除验证琼脂糖凝胶电泳图
Fig.4 The agarose gel electrophoresis ofGAL80gene knockout verification
2.4 酿酒酵母工程菌株构建及摇瓶发酵
本研究的目的是增加细胞质中Acetyl-CoA的通量,以生产目标产物异戊二烯,所以我们选择敲除有关丙酮酸和Acetyl-CoA往线粒体转运的相关基因,并在之前研究的基础上[14],选择了MPC2,POR2,PDA1基因进行敲除。将表达载体 pESC-His-ispS分别转入以上3个敲除体中,获得工程菌 SC1、SC2 和 SC3。然后摇瓶发酵来检测敲除MPC2,POR2,PDA1对异戊二烯产量的影响。结果如图5所示,3种基因的敲除都使得异戊二烯的产量有了一定的提高,其中敲除PDA1的工程菌效果最好,产量达到35 μg/L。相比较原始菌株,产量提高了0.3倍。这与CARDENAS等[14]的实验结果一致,通过敲除MPC2,POR2,PDA1会增加细胞质中Acetyl-CoA的通量。从而使目的产物的产量增加。但本研究中异戊二烯的产量没有较大提高,可能是由于细胞质中的Acetyl-CoA量还是较少,因此为了进一步增加酵母细胞质中Acetyl-CoA的通量,我们在△PDA2基础上过表达基因LmPK和CkPTA[26],获得敲除体SC4,将表达质粒 pESC-His-ispS转入SC4,获得工程菌pHB1。摇瓶发酵结果显示,过表达基因LmPK和CkPTA,异戊二烯的产量没有得到进一步的提高(图5)。因此我们推测可能是下游代谢通量较低,使得Acetyl-CoA的利用率下降。故根据上述实验结果,选取pHB1作为下一步实验的基础。

图5 敲除Acetyl-CoA支流的有关基因和引入外源基因 对异戊二烯产量的影响
Fig.5 Effects of knockout of genes related to acetyl-CoA tributaries and introduction of foreign genes on isoprene production
2.5 下游通量的验证
IDI1基因是异戊二烯合成途径中的下游基因,它编码类异戊二烯基焦磷酸异构酶,该酶参与可逆的异构化反应实现 IPP 与 DMAPP 的相互转化,是控制异戊二烯合成的关键步骤,调控该酶的表达对异戊二烯合成具有重要作用[27]。根据上述发酵实验筛选得到工程菌pHB1,我们又在pHB1菌株中过表达大肠杆菌的IDI1基因。摇瓶发酵结果如图6所示,在上述实验的基础上进一步过表达IDI1基因,使得异戊二烯的产量达到54 μg/L,比原始菌株提高了约1倍。实验结果和我们的推测一致,异戊二烯产量没有提高的原因是下游代谢通量不畅。通过对发酵液OD600进行测定,发现过表达基因LmPK、CkPTA和IDI1对菌体的生长有一定的提高,可能是代谢流调节对菌体生长有一定的促进作用。综上,通过基因敲除和过表达基因来单纯的提高细胞质中Acetyl-CoA的通量,由于代谢流的原因,并不能大幅提高异戊二烯的产量,因此还需要结合下游途径关键酶的表达来共同起作用。

图6 下游通量的验证
Fig.6 Verification of the downstream flux
3 结论
异戊二烯是重要的平台化合物,因此对异戊二烯生产方法的探索将会不断持续下去。目前利用微生物法合成异戊二烯已有诸多报道[18, 28-29],酿酒酵母作为食品安全级微生物,具有其独特的优势。本文以酿酒酵母CEN.PK2-1C为宿主菌株,引入来源于Populusalba的异戊二烯合成酶,然后敲除了Acetyl-CoA通往线粒体支流的基因MPC2、PDA1和POR2,使得酿酒酵母中异戊二烯的产量得到了一定程度的提高。然后我们又过表达了来自Leuconostocmesenteroide和Clostridiumkluyveri的木酮糖-5-磷酸特异性磷酸转酮酶基因(LmPK)和磷酸转乙酰酶基因(CkPTA)来进一步增加细胞质Acetyl-CoA的通量[26],结果表明异戊二烯的产量并没有进一步提高,我们推测是由于下游的代谢通路不畅造成的,因此我们通过过表达来源于大肠杆菌的关键酶(IDI1)进行验证,发酵结果显示,异戊二烯的产量达到了54 μg/L,比原始菌株提高了约1倍,和我们推测的基本吻合。本研究旨在探索酿酒酵母中提高异戊二烯产量的基因策略,并取得了一些成效,虽然在产量上和LV 等[11]还有较大差距,但研究为后续异戊二烯的研究工作提供了一些见解和参考,同时本研究也为异戊二烯的工业化生产奠定了一定的基础。
猜你喜欢
河南水利与南水北调(2022年7期)2022-08-18
海洋通报(2022年2期)2022-06-30
农业灾害研究(2022年1期)2022-05-07
酿酒科技(2021年8期)2021-12-06
暴雨灾害(2021年2期)2021-04-02
军事文摘·科学少年(2021年1期)2021-02-04
酿酒科技(2020年7期)2020-12-19
名人传记·财富人物(2017年9期)2017-11-02
名人传记·财富人物(2017年9期)2017-11-02
Coco薇(2016年8期)2016-10-09
