
闽楠转录组分析及基因功能注释
2020-07-14 09:03曾钦朦陈世品
植物研究 2020年4期
刘 丹 曾钦朦 刘 斌 李 煜 陈世品
(福建农林大学林学院,福州 350002)
闽楠(Phoebebournei(Hemsl.) Yang)俗称楠木,为樟科(Lauraceae)楠属(Phoebe)植物,是我国珍贵的用材树种之一,具有较高的经济和生态价值[1]。天然闽楠现存野生资源日益减少,且受到其自身生物学特性影响,生长发育缓慢,在遗传育种方面也大多采用传统育种方法,周期长且效率低[2~4]。研究闽楠群体遗传多样性,不仅可以避免物种灭绝,还可以为遗传群体的管理提供参考。目前为止,闽楠遗传多样性的研究相对较少,仅有李娟等[5]对闽楠5个野生群体进行ISSR分析,利用10对引物对96个样本的遗传多样性和遗传结构进行研究,得出闽楠在基因水平上遗传多样性较高,种群间遗传变异强于种群内。江香梅等[6]利用RAPD分子标记对福建和江西两省的8个天然群体进行遗传多样性分析,得出其遗传变异主要存在于种群内,并提出通过种群扩繁和回归自然的方式增强种群间的基因流,以维持群体遗传多样性水平。通过转录组测序开发分子标记,可以研究闽楠的群体遗传多样性,对闽楠群体管理和种质资源保护具有重要意义,也可以用于闽楠分子标记辅助育种研究,获得的转录组信息将为关键基因功能的研究提供基础。
基于二代测序技术的转录组测序技术(RNA-seq),具有高准确性、快速和低成本等优势,能够在缺乏基因资源的条件下获得物种的生长和代谢规律,并揭示其生物学特性与基因内在关联,同时可获得物种绝大多数的转录产物。通过转录组测序开发的分子标记与传统标记相比,成本低廉,简单迅速,多态性高且在植物物种间可转移,不仅可以大大提高工作效率,还可以对基因结构与功能进行更深层次的分析与探究,是一种性价比高的基因序列研究手段[7~9]。目前,转录组测序技术已经在林木遗传育种中得到广泛的应用。例如:李清莹[10]等通过对火力楠进行转录组测序,获得了97 503个Unigene基因;田晓明[11]等对半枫荷叶片进行转录组测序,共获得93 602条Unigenes,比对鉴定获得19 279个基因,经KEGG分析,有16 621个Unigenes参与了131条代谢通路;赵德刚[12]等对杜仲雌雄株进行转录组测序,获得平均长度为288 nt的雌株159 434个Unigene片段和平均长度为231 nt的雄株257 288个Unigene片段,共有48 761个表达序列标签(EST)。
目前国内有关楠属开展了浙江楠(P.chekiangensis)[13];桢楠(P.zhennan)[14];紫楠(P.sheareri)[15];白楠(P.neurantha)[16]的转录组测序分析研究工作,但未见闽楠基因组和转录组的相关报道,且未发现其他楠属树种开发的分子标记在闽楠上进行过通用性验证,遗传信息匮乏。为此,结合闽楠优质木材的特性,选取3个重要部位的组织,包括木质部、韧皮部和叶片,使用二代高通量测序技术对其进行转录组测序,获取大量闽楠转录本信息,利用生物信息分析从海量的转录本数据中筛选候选基因,以期初步了解闽楠基因表达与分布情况,为深入开展闽楠遗传育种及分子生物学相关研究奠定基础。
1 材料与方法
1.1 材料
2017年6月,在福建农林大学校园内选取1株约40年生的闽楠作为试验植株,该植株生长健康、树干通直、长势良好且无病虫害。采集试验植株的木质部、韧皮部、叶片,洗净后经液氮快速冷冻,于-80℃冰箱中保存备用。
1.2 方法
1.2.1 总RNA提取
分别使用PureLinkTMPlant RNA试剂盒(Invitrogen)提取植物总RNA,再用RQ1 RNase-Free DNase(Promega)去除总RNA中的杂质DNA。采用1.25%琼脂糖凝胶电泳检测RNA完整性,利用Agilent 2100核酸蛋白检测仪(Agilent,Santa Clara,CA)检测3个RNA样品的浓度和完整度。
1.2.2 文库构建及测序
通过Oligo(dT)磁珠富集总RNA中带有polyA结构的mRNA,采用离子打断的方式,将mRNA打断到200~300 bp片段。以mRNA为模板,用6碱基随机引物和逆转录酶合成cDNA第一链,并以第一链cDNA为模板进行第二链cDNA的合成,第二链cDNA合成时,其中的碱基T被替换成U,从而达到链特异性文库的目的。PCR扩增富集文库片段,扩增完成后进行文库片段大小的选择,文库大小在300~400 bp。通过Agilent 2100 Bioanalyzer对文库进行质检,再进行文库总浓度检测及有效文库浓度检测。采用第二代测序技术(Next-Generation Sequencing,NGS),基于Illumina HiSeq测序平台,对这些文库进行双末端(Paired-end,PE)测序。
1.2.3 数据分析
利用FastQC软件(http://www.bioinformatics.babraham.ac.uk/projects/fastqc)进行测序数据的质量评估,采用Cutadapt[17](Version 1.2.1)去除3′端的接头序列,使用Trinity[18](版本r20140717,K-mer 25 bp)软件进行转录组De Novo组装,利用转录组表达定量软件RSEM[19]计算每个基因的FPKM值,采用DEGSeq[20]分析表达差异Unigene,用Varscan[21](Version 2.3.7)程序获取cSNP位点。使用MISA(http://pgrc.ipk-gatersleben.de/misa/misa.html)程序在序列中搜索SSR位点。
2 结果与分析
2.1 测序数据过滤
将RNA片段化,建立PCR富集文库,文库插入长度为380 bp,采用Illumina MiSeq测序平台测序,测序模式为双末端的2×150 bp(见表1)。测序过滤后的数据平均质量较好,可以进行后续分析。
2.2 转录本拼接结果
提取每个基因下最长的转录本作为该基因的代表序列,称为Unigene。对拼接得到的Contig、Transcript和Unigene序列进行统计(见表2)。
2.3 Unigene功能注释结果
对聚类得到的Unigene进行基因功能注释(见表3)。基因功能注释所用到的数据库包括NR(NCBI non-redundant protein sequences)、GO(Gene Ontology)[22]、KEGG(Kyoto Encyclopedia of Genes and Genome)[23]、eggNOG(evolutionary genealogy of genes:Non-supervised Orthologous Groups)[24]、Swiss-Prot。由表3可知,NR数据库注释了45 036条Unigene,获得的信息最多,KEGG数据库获得的信息最少,仅有5 623条Unigene得到注释,在所有数据库中均被注释到的Unigene有4 646条,仅占3.06%。

表1 测序数据统计
注:Q30(bp):碱基识别准确率在99.9%以上的碱基总数;N(%):模糊碱基所占百分比;Q20(%):碱基识别准确率在99%以上的碱基所占百分比;Q30(%):碱基识别准确率在99.9%以上的碱基所占百分比;GC(%):GC含量
Q30(bp). The total number of bases whose recognition accuracy is above 99.9%; N(%). Percentage of fuzzy bases; Q20(%). The percentage of bases whose recognition accuracy is above 99%; Q30(%). The percentage of base recognition accuracy above 99.9%;GC(%). GC content

表2 转录本拼接结果
注:N50(bp).将所有序列从长到短排列,将序列长度按照该顺序依次相加,当相加的长度达到序列总长度的50%时,最后一条序列的长度;N90(bp).将所有序列按照长度从长到短排列,将序列长度按照该顺序依次相加,当相加的长度达到序列总长度的90%时,最后一条序列的长度;N50 Sequence No..长度大于N50的序列总数;N90 Sequence No..长度大于N90的序列总数;GC(%).序列的GC含量
Note:N50(bp).All sequences are arranged from long to short,and the sequence lengths are added in this order. When the added length reaches 50% of the total sequence length,the length of the last sequence is; N90(bp).All sequences are arranged from long to short in length,and the sequence length is added in this order. When the added length reaches 90% of the total sequence length,the length of the last sequence is; N50 Sequence No..Total number of sequences longer than N50; N90 Sequence No..Total number of sequences longer than N90; GC(%).The GC content of the sequence
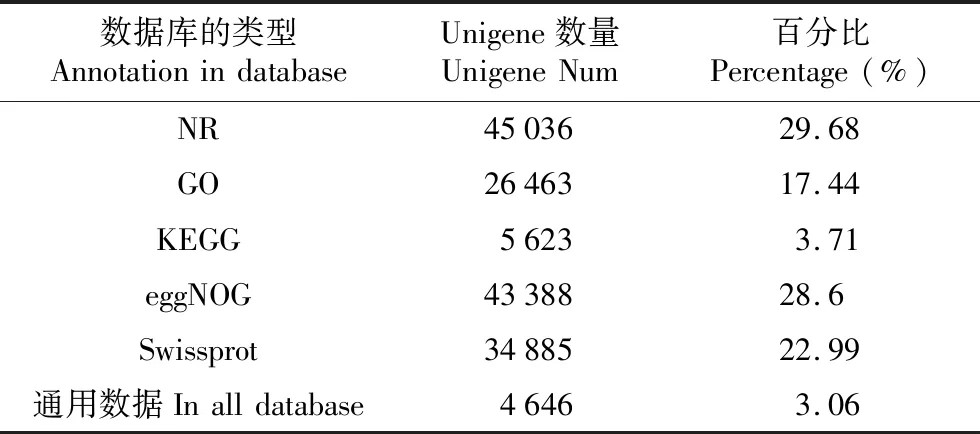
表3 Unigene功能注释结果

图1 NR注释结果统计图Fig.1 NR annotated species distribution of Unigenes of transcriptome for P.bournei
2.3.1 闽楠转录组Unigene的NR功能分类
通过Blastx将闽楠转录组中的Unigene与NR数据库进行比对,可以获取本物种基因序列与近缘物种基因序列的相似性以及本物种基因的功能信息,其中有45 036条Unigene有注释结果。通过NR库比对注释的结果,统计并绘制比对上的物种分布图(见图1)。闽楠与黄瓜、野草莓、大豆、欧洲大叶杨、碧桃、蓖麻、葡萄都有同源序列分布,其中与葡萄有34%的同源序列,与欧洲大叶杨、碧桃、蓖麻分别有8%、7%、6%的相似序列,在黄瓜、野草莓、大豆中的同源序列较少,均为3%。此外,还有35%的Unigene属于其他序列,可能包含了闽楠自身特有的与大多数物种不同的序列。

表4 闽楠转录组GO功能分类的Unigene数量分布

图2 闽楠转录组Unigene的GO功能分类Fig.2 GO analysis of P.bournei transcriptome Unigene
2.3.2 闽楠转录组Unigene的GO分类注释
对闽楠转录组Unigene进行GO功能分类,有26 463条Unigene获得了基因注释,占总Unigene数的17.44%。将注释的到的Unigene划分为生物过程、细胞成分、分子功能3大类共计52个分支(见图2),对每一类的基因数量进行统计发现(见表4),在生物过程这一类中,代谢过程、细胞过程、单一有机体过程占比较大;在细胞成分一类中,细胞、细胞器、膜、细胞组分占比较大;在分子功能一类中,催化活性和连接占比较大。
2.3.3 闽楠转录组Unigene的eggNOG功能注释
将闽楠转录组的Unigene与eggNOG数据库进行比对,可得到43 388个Unigene具有功能信息,根据功能可将其划分为25类(见图3:A~W,Y,Z)。由图3可知,一般功能预测(R)的Unigene数量最多,其次为未知功能(S)和信号传导机理(T),细胞动力(N)最少,仅有7条Unigene注释。
通过对每一类eggNOG注释的基因功能进行统计,可获得闽楠转录组中各类功能基因的数量分布信息(见表5)。其中,此生代谢产物生物合成、运输及代谢(Q)有1 617条Unigene。
2.3.4 闽楠转录组Unigene的KEGG代谢途径分类分析
根据KEGG数据库的注释信息进一步将闽楠Unigene进行pathway注释,共有5 623条Unigene获得注释,参与的代谢通路可归为5个大类、35个子类,又可分为176个信号代谢分支(见图4)。其中,新陈代谢(Metabolism)一类中获得注释最多的是碳代谢(Carbon metabolism),有164条Unigene;遗传信息处理(Genetic Information Processing)一类中核糖体(Ribosome)获得注释最多,有221条Unigene;环境信息处理(Environmental Information Processing)一类中获得注释最多的是植物激素信号转导(Plant hormone signal transduction),有118条Unigene,细胞进程(Cellular Processes)和有机系统(Organismal Systems)两类中获得注释最多的分别为内吞作用(Endocytosis)96条、生成信号通路(Neurotrophin signaling pathway)90条(见表6)。
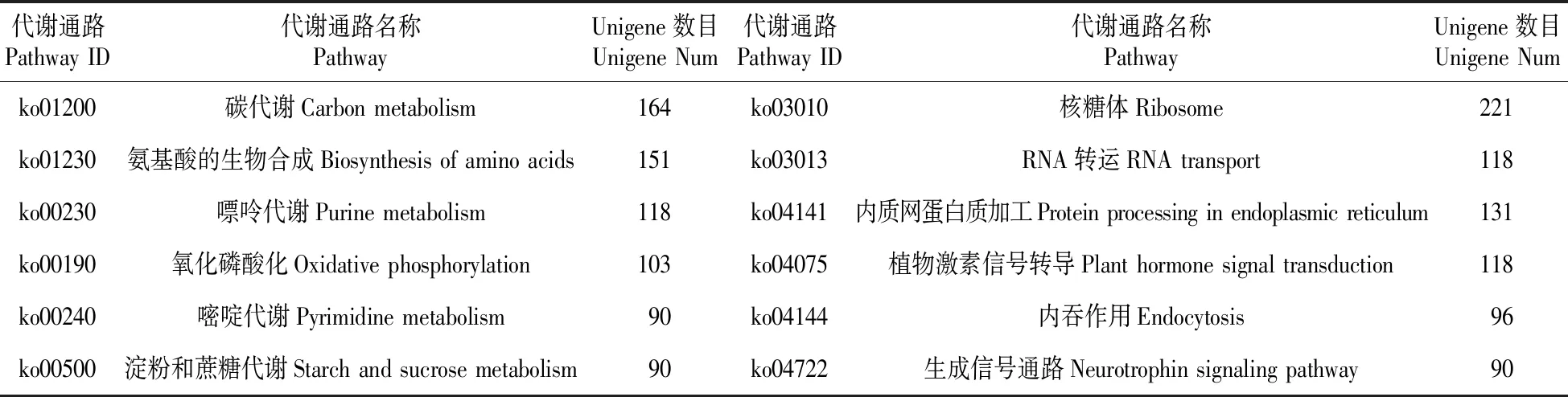
表6 闽楠转录组的KEGG代谢途径(>1.5%注释基因)

图3 闽楠转录组Unigene的eggNOG功能分类 A.RNA加工与修饰;B.染色质结构与动力;C.能量生成与转化;D.细胞周期调控,细胞分裂,染色体分配;E.氨基酸运输及代谢;F.核苷酸运输及代谢;G.糖类运输及代谢;H.辅酶运输及代谢;I.脂类运输及代谢;J.翻译,核糖体结构和生物合成;K.转录;L.复制,重组与修复;M.细胞壁生源/细胞膜生源/囊膜生源;N.细胞动力;O.翻译后修饰,蛋白质转换与分子伴侣;P.无机离子的运输及代谢;Q.次生代谢产物生物合成,运输及代谢;R.一般功能(预测);S.未知功能;T.信号转导机理;U.胞内运输,分泌及小泡运输;V.防御机制;W.胞外结构;Y.细胞核结构;Z.细胞骨架Fig.3 eggNOG analysis of P.bournei transcriptome Unigene A. RNA processing and modification; B. Chromatin structure and dynamics; C.Energy production and conversion; D. Cell cycle control,cell division,chromosome partitioning; E. Amino acid transport and metabolism; F. Nucleotide transport and metabolism; G.Carbohydrate transport and metabolism; H. Coenzyme transport and metabolism; I. Lipid transport and metabolism; J.Translation,ribosomal structure and biogenesis; K.Transcription; L. Replication,recombination and repair; M.Cell wall/membrane/envelope biogenesis; N. Cell motility; O.Posttranslational modification,protein turnover,chaperones; P. Inorganic ion transport and metabolism; Q.Secondary metabolites biosynthesis,transport and catabolism; R.General function prediction only;S.Function unknown;T.Signal transduction mechanisms;U.Intracellular trafficking,secretion,and vesicular transport; V.Defense mechanisms; W.Extracellular structures;Y.Nuclear structure; Z.Cytoskeleton
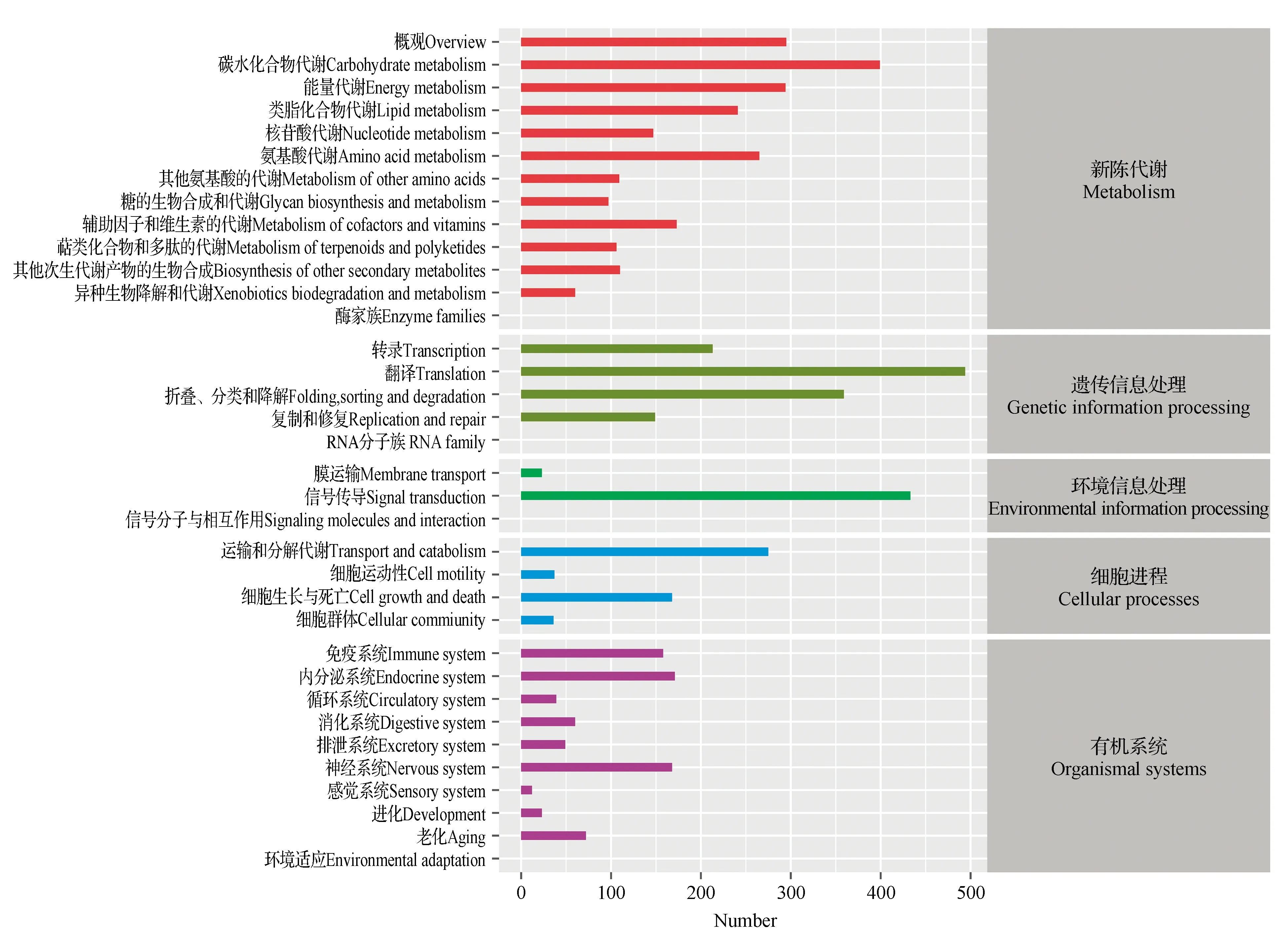
图4 KEGG注释统计图Fig.4 KEGG annotation statistical chart

表7 闽楠转录组SSR搜索结果
2.4 闽楠转录组Unigene的SSR分布特征
SSR(Simple Sequence Repeats),称为短串联重复序列或微卫星标记[25],通过对闽楠的151 729条Unigene进行SSR位点搜寻,共检测到35 972个SSR位点,SSR重复类型从单核苷酸到六核苷酸均有不同数量的分布,所占比例有较大差异,其中单核苷酸数量最多,占60.5%,六核苷酸仅占0.04%(见表7)。
3 讨论
本研究对闽楠的木质部、韧皮部、叶片3个不同部位的组织进行高通量测序,获得木质部41 383 707条、韧皮部43 343 922条、叶片44 191 586条Clean Reads片段,经转录本拼接后得到序列总长度达120 535 288bp的383 331条Conting片段,进一步组装得到平均长度为542 bp的151 729条Unigenes。相比于其他利用相同测序手段的物种来说长度较长,如茶(Camelliasinensis)(平均长度355 bp)[26]、杉木(Cunninghamialanceolata)(平均长度449 bp)[27]、枣(Ziziphuscelata)(平均长度408 bp)[28]等。在Swiss-Prot数据库中共检测到34 885条蛋白同源序列,与NR数据库进行比对发现,与葡萄科葡萄属的葡萄(Vitisvinifera)比对的同源信息最多,占34%,出现此种情况,可能是由于葡萄与闽楠的进化史和生活史较为相似,也可能是因为葡萄具有参考基因组;与黄瓜、野草莓、大豆等物种相比同源性较低,仅有3%,另有35%的Unigene未在数据库中找到同源序列,可能是因为闽楠本身特有的基因存在,也可能是因为这些序列是一些非编码的RNA序列,或是因为长度较短不包含蛋白质功能域信息,也有可能是目前公共数据库中的基因信息还不够完善[29]。从进化关系上来看,不同科属的植物,其具有同源序列的数量较低,闽楠符合这一特点。将闽楠转录组Unigene注释到GO数据库,可划分为生物过程、细胞成分、分子功能3大类共计52个分支,与eggNOG数据库比对可分为25类,通过KEGG功能注释可知转录组中涉及的基因共参与了176条代谢通路,其中涉及代谢通路和次生物质的生物合成基因较多。此外,转录组序列中共检测到35 972个SSR位点,其中单核苷酸占比最大,达到60%以上。
丁亚军[13]等在2014年对浙江楠叶、木质部、韧皮部、花、花柄5个组织的转录组进行测序,共获得了52 527 954条Reads片段,经组装拼接后得到平均长度为711.25 bp的111 250个Unigenes,在NR、Swiss-Prot、GO、KEGG等数据库中均获得较好的注释。有3大类60个分支的36 370条Unigenes成功注释到GO数据库,有25类15 605条Unigenes具有具体的蛋白功能定义,16 135条Unigenes在KEGG数据库映射到298条代谢通路中。值得注意的是,在与NR数据库比对中发现,与浙江楠的Unigene同源信息最高的是葡萄科葡萄树的葡萄,占比62%,其次为杨柳科的毛果杨和铁苋菜亚科的蓖麻,各占12%,与豆科的青仁乌豆和苜蓿共有8%相似,这与闽楠在NR数据库中的同源序列比对结果有一定的相似性,但也存在差异。由此可见,闽楠和浙江楠虽同为樟科楠属植物,但是在基因的同源性分布上仍有不同,一方面可能是因为转录组测序结果的时空异质性较高,不仅不同物种差异较大,同物种,乃至同植株在不同阶段的测序差异结果也会较大,我们在转录组测序时用于提取RNA选取的组织来自于不同部位,导致了不同组织器官在不同时期具有不同的基因表达,另一方面可能是因为与其他物种进行比对具有一定的局限性。
本研究通过生物信息学方法对闽楠进行转录本拼接、基因功能注释、SNP检测以及SSR位点预测等,获得的转录组数据不仅可以作为樟科楠属树种遗传信息的重要组成部分,进一步丰富该科树种的基因数据库,同时也为闽楠基因的分子克隆和功能鉴定提供数据支持,除了可以发掘候选基因外,还可以开发EST-SSR分子标记,为后续闽楠的遗传多样性分析奠定基础。
猜你喜欢
湘潮(上半月)(2022年7期)2022-12-06
中国人兽共患病学报(2022年9期)2022-10-19
科学导报(2021年29期)2021-06-03
猪业科学(2021年3期)2021-05-21
科学之谜(2021年2期)2021-04-25
中国生殖健康(2020年4期)2021-01-18
少先队活动(2020年12期)2021-01-14
科学导报(2020年54期)2020-09-09
中华诗词(2019年1期)2019-11-14
学苑创造·B版(2019年5期)2019-06-14
