
婴儿期起病的甲基丙二酸血症41例病例系列报告
2021-01-28 00:54邓冬立肖非凡吴冰冰孙卫华周文浩
中国循证儿科杂志 2020年6期
邓冬立 肖非凡 吴冰冰 孙卫华 杨 琳 陆 炜 周文浩
甲基丙二酸血症(MMA)又称甲基丙二酸尿症,是一种罕见的先天性有机酸代谢异常导致多系统损伤的疾病,为常染色体隐性遗传病,于1968年被首次报道[1]。MMA是中国患儿最常见的先天性有机酸代谢性疾病, 发病率为1/250 000~1/48 000[2, 3]。甲基丙二酸正常情况下在甲基丙二酰辅酶A(MCM)及其辅酶钴胺素(cbl,即维生素B12)的作用下转化生成琥珀酸,参与三羧酸循环。MCM缺陷或cbl代谢异常可导致甲基丙二酸等代谢物蓄积,引起神经系统、肾脏等多系统损伤。目前研究[4]显示,婴儿期发病的MMA起病症状多样,预后较差,但及时诊治可以改善预后。本文回顾性分析复旦大学附属儿科医院(我院)分子医学中心通过基因检测确诊的婴儿期发病的MMA的临床资料和基因突变情况,促进早期诊断和干预。
1 方法
1.1 MMA的诊断标准 质谱检测提示MMA,或基因检测证实存在MMA相关基因致病变异。
1.2 病例纳入标准 ①2016年6月至2019年6月在我院分子医学中心行基因检测,发现MMA已知致病基因(MUT、MMACHC、MMAA、MMAB、MCEE、MMADHC、LMBRD1、ABCD4、PRDX1、HCFC1)的致病或可疑致病变异;②诊断年龄<1岁。
1.3 高通量基因检测 在取得患儿父母的知情同意后,采集患儿外周静脉血。用mini blood全血试剂盒(QIAGEN公司,德国)提取基因组DNA,NanoDrop 2000(Thermofisher公司,美国)紫外分光光度仪测定其浓度。基于Illumina Hiseq 2000/2500 平台进行序列检测。测序覆盖度>98%,平均测序深度>100×。根据我院高通量测序数据分析流程 (第2版)[5]对变异进行信息注释和致病性分析。
1.4 资料截取 从我院病历系统中获取以下信息:①患儿的性别、胎龄、出生体重、发病年龄、分娩方式、家族史;②根据OMIM数据库对MMA的表型记录,重点收集整理就诊时主要临床表现:发育延迟、肌张力异常、喂养困难、肝损伤、呕吐、神经影像学改变、惊厥、心肌受累、呼吸系统异常、低钾、血液学异常、酸中毒、肾脏受累、骨骼异常;③高通量基因检测结果。
1.5 统计学分析 采用SPSS 20.0软件。计数资料用卡方检验或Fisher确切概率法进行比较。P<0.05为差异有统计学意义。
2 结果
2.1 一般情况 41例婴儿期起病的MMA患儿进入本文分析,男26例,女15例;中位起病时间为21日龄(3日龄至1岁),男、女患儿中位起病时间分别为18和24日龄。新生儿期就诊25例,~6月龄就诊12例,~12月龄就诊4例。主要就诊原因包括:生后反应差24例,遗传代谢病串联质谱筛查疑似MMA 8例(其中2例同时有同型半胱氨酸增高)。发育迟缓3例,抽搐2例,气促伴呕吐、单纯气促、肺动脉高压、四肢青紫各1例。
2.2 基因检测结果 41例共检出2个基因47种变异。
25例携带MUT基因致病/可疑致病变异,2例为纯合变异,其余均为复合杂合变异。共检测到31个不同的变异位点,变异类型为:移码突变9个、错义突变28个、剪接供体突变4个、终止突变7个。热点致病变异为c.729_730insTT(5例)、c.323G>A(4例)、c.1677-1G>A(4例)。
16例携带MMACHC基因致病/可疑致病变异,均为复合杂合变异。检测到16个不同的变异位点,变异类型:移码突变8个、框内缺失突变3个、错义突变6个、剪接供体突变1个、启动子丢失突变1个、终止突变13个。热点致病变异:c.609G>A(8例)、c.567dupT(5例)、c.80A>G(4例)。
2.3MUT和MMACHC基因缺陷导致MMA的临床表型比较 8例为新生儿质谱筛查怀疑MMA,尚无临床表型,不纳入基因型-临床表型比较分析。其余33例中,22例携带MUT基因变异(MUT组),11例携带MMACHC变异(MMACHC组)。
MUT组,男16例、女6例;新生儿期就诊17例,~6月龄就诊2例,~12月龄就诊3例;中位起病年龄为11日龄。MMACHC组,男7例,女4例;新生儿期就诊5例,~6月龄就诊5例,~12月龄就诊1例;中位起病年龄为2月龄。
表1显示,2组就诊主要原因差异无统计学意义;2组最常见的表型均为肌张力异常、呼吸系统异常和喂养困难,差异无统计学意义。MUT组更易出现酸中毒(50.0%vs9.1%,P=0.027),MMACHC组更易出现心肌受累表型(36.4%vs4.5%,P=0.037)。此外,MUT组6例患儿行MR检查,结果提示1例正常、1例代谢性脑病、2例脑外隙增宽、2例皮下血肿;3例患儿行CT检查,结果提示1例正常、2例脑白质密度减低。MMACHC组5例患儿行MR检查,结果提示1例正常、1例脑积水、1例白质髓鞘化落后、2例脑外隙增宽;1例患儿行CT检查,结果提示脑积水。
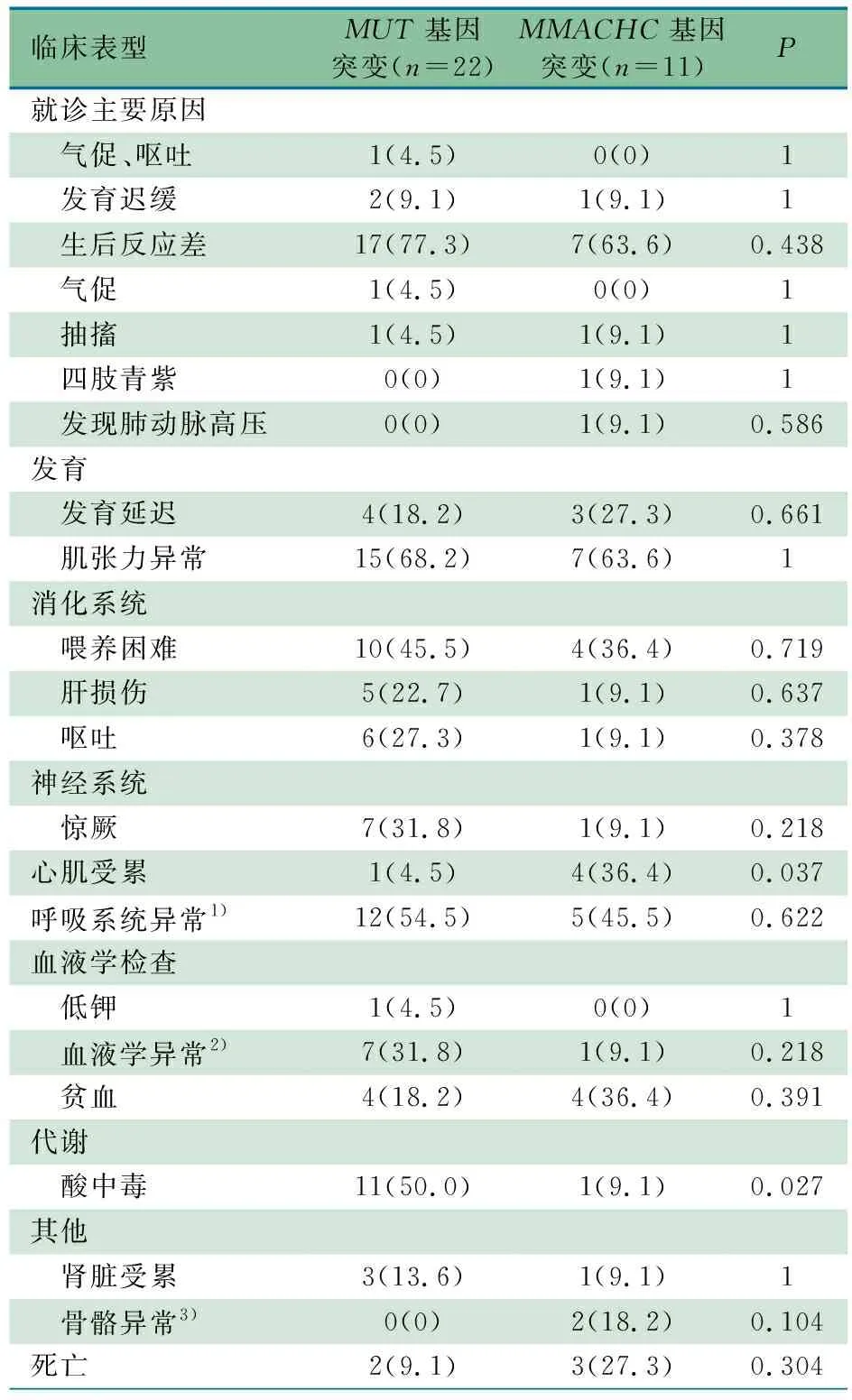
表1 MUT和MMACHC基因缺陷致MMA临床表型比较[n(%)]
2.4 死亡患儿分析 5例患儿放弃治疗后死亡,包括多脏器功能衰竭3例,脑疝和严重脑积水各1例。2例携带MUT基因的c.626dupC/c.787G>A和c. 729_730insTT/c.1679G>A,2例携带MMACHC基因的c.609G>A/c.658_660delAAG变异,1例携带MMACHC基因的c.80A>G/c.567dupT变异。
3 讨论
MMA常见于新生儿和婴幼儿,因其临床表现缺乏特异性而容易误诊,得到及时诊治者预后较好。本研究41例<1岁起病的MMA患儿中,仅8例(19.5%)为新生儿期尚无临床表现前,串联质谱筛查提示MMA;其余33例因有临床表现就诊的患儿中,最主要的就诊原因为反应差。如就诊时合并肌张力异常、喂养困难,应高度怀疑MMA可能性。但上述症状在婴幼儿期特异性差,而高通量测序技术的发展为MMA的早期诊断提供了极大的便利。有研究报道,中国MMA患儿最常见的变异基因为MMACHC基因和MUT基因[6]。本文对41例婴儿期起病的MMA患儿的基因检测结果与之一致, 25例为MUT基因变异,16例为MMACHC基因变异。
MUT基因位于染色体6p12.3区域,包含13个外显子,全长约35 kb,其编码的MCM酶主要有2个功能结构域[7],N-末端的结构域(第88~422位氨基酸)为底物结合区,C-末端的结构域(第578~750位氨基酸)属于cbl结合区。本文中MUT基因变异位点主要位于N-末端结构域的第2、3、6外显子,可引起蛋白质二级结构的改变,从而影响MCM酶活性。Liu等[8]报告,37.5%的MUT基因突变位点位于第3、6外显子。因此,MUT基因的第3、6外显子为突变的热点区域,机制尚不明确。MMACHC基因位于染色体1p34区域,含4个外显子,编码cblc蛋白。该基因发生突变导致cblc蛋白缺陷,破坏了正常的氰钴胺的还原脱氰反应,引起机体甲基丙二酸及同型半胱氨酸蓄积。本文MMACHC变异多聚集在外显子4区域,与此前报道[9]一致,提示该区域为MMACHC变异热点区域。
目前已知的中国MMA患者MUT基因热点变异主要为c.729_730insTT、c.1106G>A、c.914T>C和c.2080C>T[10],MMACHC基因热点变异主要为c.609G>A、c.80A>G和c.658_660 delAAG[9, 11]。本文MUT基因常见热点变异为c.729_730insTT、c.323G>A和c.1677-1G>A,MMACHC基因常见热点变异为c.609G>A、c.567dupT和c.80A>G。本文与文献报道的热点变异不完全一致,可能与患儿来源不同有关。文献[9~11]以中国北方人群为主,本文患儿主要来自中国南方地区。本研究中检测到的最常见的MMACHC基因突变位点为c.609G>A,该突变为无义突变,可导致色氨酸编码提前终止,从而引起cblc蛋白缺陷,也是我国MMACHC基因的热点突变[12]。
MMA患儿常见的临床表现包括呕吐、喂养困难、惊厥和发育迟缓等,本文患儿就诊主要原因与之较一致。本文病例中,携带MUT基因突变的患儿较携带MMACHC基因突变者更易出现酸中毒症状,与Kang等[13]的报告一致。有研究[14]显示,出现酸中毒表现的MMA患儿后期易出现神经系统异常及其他长期并发症。本文还发现,携带MMACHC基因变异的患儿更易有心肌受累的表现。心肌病是MMA较为罕见的症状,Kang等[13]报告的301例MMA患者中7例(2.3%)有心肌病表现,He 等[15]报告的132例携带纯合MMACHCc.609G>A变异的患儿均无心肌病表现,白薇等[16]报告10例有心血管系统受累的MMA患儿,9例有MMACHCc.80A>G纯合/杂合变异。本文中携带MMACHC基因变异的患儿36.4%有心肌受累表现,均为c.80A>G杂合变异。推测MMA患儿心肌受累表型可能与c.80A>G杂合/纯合变异相关。Tanpaiboon等[17]提出,宫内MMACHC酶缺乏可能是引起患儿心脏发育异常的原因。分子机制研究[18,19]也表明,MMACHC基因表达异常可引起细胞超微结构改变和线粒体功能障碍,进而诱发心脏发育异常。此外,本文发现1例携带MUT基因c.1208G>A/c.1286A>G变异的患儿存在心肌受累的表型。但是MUT基因变异导致心肌受累的文献报道较少,仍需进一步的分子机制研究。
本文2例携带复杂MUT基因变异的死亡患儿均携带移码突变。MUT基因的c.626dupC变异和c. 729_730insTT变异均位于C-末端的结构域上,可影响钴胺素结合,从而引起蛋白质构象改变。先前研究报道中国携带MMACHC基因的c.609G>A/c.658_660delAAG突变位点的患儿均发病早、症状重、预后差[20]。本文3例死亡患儿中,2例携带MMACHC基因c.609G>A/c.658_660delAAG变异,1例携带MMACHC基因c.80A>G/c.567dupT变异。推测c.609G>A突变结合移码变异可导致较为严重的临床表型。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国食物与营养(2022年5期)2022-06-17
河北果树(2021年4期)2021-12-02
实用临床医药杂志(2021年13期)2021-08-07
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年4期)2021-01-18
医药前沿(2020年20期)2020-11-10
当代畜禽养殖业(2020年9期)2020-10-21
家庭医学·下半月(2020年2期)2020-04-26
科学生活(2019年7期)2020-01-01
