
Ni、Mo共掺杂对SnO2材料性能影响的仿真研究
2021-02-23 12:29常永强王景芹朱艳彩张广智胡德霖
人工晶体学报 2021年1期
常永强,王景芹,朱艳彩,张广智,胡德霖
(1.河北工业大学,省部共建电工装备与智能化国家重点实验室,天津 300130;2.上海良信电器股份有限公司,上海 200137;3.苏州电器科学研究院有限公司,苏州 215000)
0 引 言
被广泛使用的电接触材料中,AgCdO因具有良好的抗熔焊性、抗电弧侵蚀性和较小的接触电阻而大量应用于各种场合,但由于Cd元素具有毒性,其生成物危害较大,目前AgCdO已经被限制使用[1]。AgSnO2与AgCdO相比,不仅电接触性能更加优越,而且生成物没有危害,故被认为是代替AgCdO的最佳材料之一。然而AgSnO2触头材料仍存在很大的不足:由于电弧的影响,促使大量的SnO2颗粒聚集在接触表面,SnO2属于宽禁带半导体,导电性很差,会使触头间的接触电阻增大;其次,SnO2本身的硬度很高,不利于AgSnO2的成型并且对后续加工造成了困难,SnO2性能上的缺陷极大地制约了AgSnO2的推广及使用[2-4]。
针对AgSnO2性能的不足,研究人员尝试通过不同的制备方法来提高触头材料的性能,如机械合金化法、化学共沉淀法、反应雾化法、预氧化合金法以及反应合成法等,这些方法大多都是通过改善SnO2颗粒在银基体中的分布情况,以此改善触头的性质,提高触头的各项性能[5]。除此之外,研究人员在经过大量的实验之后发现,在AgSnO2中添加微量元素也可以明显改善AgSnO2触头材料的各项性能。康慧玲等[6]通过Materials Studio软件建立SnO2模型,对Sr单掺和Sr-F共掺后的SnO2模型进行优化计算,从理论上分析了掺杂后SnO2的导电性能,为AgSnO2材料的发展和研究提供新的思路和方法。张颖等[7]在SnO2中添加La、Ce和Y元素并计算了掺杂前后的弹性常数,发现Y掺杂SnO2的普适弹性各向异性指数最小,触头产生裂纹的概率减小,电寿命增加,且Y掺杂可以明显减小SnO2的硬度,使得触头材料加工和成型更加容易[7]。
近年来,高价金属掺杂SnO2受到人们的广泛关注与研究,高价金属Mo掺杂制得的SnO2薄膜具有良好的显微结构以及表面性能[8-10]。AgNi触头在发生电弧时,弧根区域的温度很高,Ni颗粒会在达到熔点后大量溶解在Ag基体中,且在短时间内迅速熔化并冷却,形成二次结晶,在Ni熔化的过程中会大量吸收电弧的能量,达到灭弧的效果,可以提高触头的抗电弧侵蚀能力,此外,Ni的硬度较低,易于加工[11]。综合Ni与Mo元素的优点,本文尝试研究Ni单掺杂、Mo单掺杂以及Ni-Mo共掺杂对SnO2材料电性能、力学性能的影响。
本文运用基于密度泛函理论(Density Functional Theory,DFT)的第一性原理方法,计算得到了未掺杂SnO2,Ni、Mo单掺杂及Ni-Mo共掺杂SnO2模型的能带结构、态密度、弹性常数等参数,通过第一性原理的计算,从理论上分析了掺杂元素对于SnO2材料导电性质、力学性质的影响,为触头材料的研发提供参考。
1 理论模型与计算方法
1.1 理论模型
SnO2晶胞是四方相金红石结构,一个SnO2晶胞由2个Sn原子和4个O原子组成,本文计算采用的1×2×3超晶胞模型中包含12个Sn原子和24个O原子,如图1(a)所示。掺杂采用原子替代的方法,单掺杂模型如图1(b)和(c)所示,SnO2超晶胞模型中相邻的两个Sn原子分别被两个Ni原子、两个Mo原子替代,共掺杂模型如图1(d)所示,SnO2超晶胞模型中相邻的两个Sn原子分别被一个Ni原子和一个Mo原子替代,单掺杂与共掺杂浓度比例均为16.67%。
1.2 计算方法
本文的各项物理参数均由Material Studio软件的CASTEP模块计算而得,采用基于密度泛函理论的平面波超软赝势方法来描述价电子与离子之间的相互作用,电子与电子之间的交换关联能选择广义近似方法(GGA)下的修正泛函(PBE)进行计算[12-13],布里渊区采用5×5×8的Monkorst-Park方案,平面波截断能选择400 eV。计算过程中,为了保证后续计算的正确性,首先对晶胞进行几何优化,使结构总能量达到最小值,找到结构最稳定点。达到稳定状态以后,进行能带、态密度、弹性常数等参数的计算,进而分析各模型的导电性质、力学性质,选用的价电子组态为:Sn 5s25p2、O 2s22p4、Ni 3d84s2、Mo 4s24p64d55s1。
2 结果与讨论
2.1 晶胞参数分析
未掺杂的SnO2,元素Ni、Mo单掺杂SnO2,Ni-Mo共掺杂SnO2经几何结构优化后的晶胞参数如表1所示。可以看出SnO2掺杂之后的晶格常数相较于未掺杂SnO2在a轴方向上有所增大,而在c轴方向上有所减少,且晶胞体积以及键长都有所减小。这是因为Sn4+的半径为0.069 nm,Ni2+的半径与Sn4+相同,为0.069 nm,而 Mo6+的半径为0.065 nm,由量子化学理论可知,Mo6+的半径小于Sn4+,Mo单掺杂后的晶胞体积应减少,但从表中数据看出,Mo单掺杂的晶胞体积与未掺杂相近,这是因为Mo原子替换Sn原子后,产生了多余的正电荷,这些正电荷相互之间的排斥作用使得晶胞体积变大[14]。同理,虽然Ni2+的半径与Sn4+相同,但是正电荷减少,使得Ni单掺杂的晶胞体积减小,Ni-Mo共掺杂模型的晶胞参数大小处于Ni单掺以及Mo单掺杂之间。

图1 SnO2及其掺杂模型

表1 晶胞参数
2.2 稳定性分析
掺杂形成能的大小能够反映掺杂模型形成的难易程度以及形成后体系的稳定性,对于能否成功制得相应的实验样品非常重要,因此本文计算了各掺杂模型的形成能,公式如下[15]:
Ef(X)=ESnO2X-ESnO2-nEX+mESn
(1)
Ef(Ni,Mo)=ESn(1-x)NixO(2-y)Moy-ESnO2-nENi+mESn-pEMo
(2)
式(1)用于计算Ni单掺杂以及Mo单掺杂的形成能,式(2)用于计算Ni-Mo共掺杂模型的形成能。式中ESnO2X表示X(Ni、Mo)元素单掺杂且经过几何优化后的SnO2体系总能量,ESnO2表示未掺杂的体系总能量,ESn(1-x)NixO(2-y)Moy表示共掺杂体系总能量,EX表示X(Ni、Mo、Sn、O)原子的基态能量,n、p、m表示为掺杂Ni、Mo原子个数以及被替换的Sn原子个数。各模型的总能量以及计算所得的形成能如表2所示,当形成能为负值时,其绝对值越大,说明此晶体越容易合成[16]。由表2数据可知,Ni单掺杂、Mo单掺杂以及Ni-Mo共掺杂的形成能均为负值,可见三种体系的稳定性都较好,且当Mo掺杂时计算所得的形成能绝对值最大,即此掺杂模型最容易形成。

表2 总能量与形成能
2.3 电子结构分析
2.3.1 能带分析
图2为未掺杂SnO2、Ni单掺杂、Mo单掺杂以及Ni-Mo共掺杂的能带结构,选择0 eV作为费米能级。由图所知,未掺杂SnO2的价带顶和导带底均位于布里渊区的G点,属于直接带隙半导体材料,带隙值为1.059 eV,小于实验值3.6 eV[17],姜如青等[18]建立了In-Ga共掺杂SnO2模型,计算未掺杂SnO2模型的带隙值为1.27 eV,丁超等[19]计算未掺杂SnO2模型的带隙值为1.039 eV,理论计算所得的带隙值普遍要比实验值低,这是由于CASTEP模块中GGA算法低估了导带中激发态电子的能量,从而导致带隙值偏小,但并不影响掺杂前后带隙值的分析[20]。由掺杂后的能带图可知,掺杂后仍为直接带隙半导体,导带和价带变得更加密集,且导带部分能带和价带部分能带均向费米能级移动,带隙值减少,电子由价带被激发到导带所需的能量变小,载流子浓度增大,导电性增强;能带间起伏趋于平缓,局域性增强。图2(b)为Ni单掺杂后的能带结构,理论分析可知,一个Ni原子代替一个Sn原子后,Ni-O成键导致有两个空穴未配对,成为自由空穴,占据费米能级以下的价带成为受主能级,形成P型掺杂半导体,且导带和价带之间出现了中间能级,使得载流子跃迁更加容易,所需能量减少,与未掺杂SnO2相比,带隙值减少为0.612 eV。同理可知,Mo单掺杂时,Mo-O成键后多于两个电子未配对,成为自由电子,占据费米能级上的导带成为施主能级,形成N型半导体,由2(c)可看出,Mo单掺杂后,费米能级出现在了导带部分,带隙值为0.618 eV。由2(d)可知,Ni-Mo共掺杂之后,导带底部穿过了费米能级,为N型掺杂,能级数目明显增多,导带和价带处的能级变得更加密集,带隙值进一步减少为0.297 eV。
2.3.2 态密度分析
未掺杂SnO2和单掺杂、共掺杂SnO2的态密度图如图3所示。
图3(a)为未掺杂SnO2的态密度图,从图中可以看出,在深能级能量(-19.5~-15.6 eV)出现峰值,主要是由于O的2s轨道提供能量,但是距离费米能级较远,对导电性影响较小。在费米能级附近价带的能量(-8.4~0.3 eV)出现峰值,主要是由O的2p轨道,以及少量Sn的5s、5p轨道提供能量。导带部分(2.1~27.7 eV)的能量主要是由Sn的5s、5p轨道同时提供。由图3(b)可知,Ni单掺杂之后,Ni的3d轨道对费米能级附近能带(-5.3~1.8 eV)的影响较大,形成的Ni-O键使轨道杂化,价带穿过了费米能级,提供大量的空穴载流子,帮助电子更容易转移,导电性增强,与能带图分析一致。图3(c)所示为Mo单掺杂的态密度图,Mo的4p、4s以及5s轨道对导带部分(6.9~20.6 eV)影响较大,导带部分的能级更加密集,费米能级附近能带(-9.2~8.3 eV)受Mo的4d轨道影响较大,Sn的5p、5s轨道与Mo的轨道相互杂化,致使导带发生下移,穿过了费米能级。从图3(d)中可以看出,Ni-Mo共掺杂后,Ni的3d轨道和Mo的4d轨道在费米能级附近(-5.8~5.7 eV)共同作用,使得能带的价带上移,导带下移,电子跃迁所需的能量减少,导电性得到了提高。
2.3.3 电荷布居分析
表3为未掺杂SnO2以及不同元素掺杂SnO2的原子电荷布居表,表中数据均为平均值。通过电荷布居数可以对晶胞内各原子的电荷转移、成键情况以及成键的强弱进行定量分析。对于未掺杂SnO2而言,Sn原子和O原子的电荷布居数分别为1.90和-0.95,Sn失电子带正电荷,O得电子带负电荷,掺杂后Sn的电正性减弱,O的电荷布居数增多,对比表中数据可知Ni的失电子能力最强。键重叠布居数可以反映成键的性质和强弱,布居数越大,原子间的成键作用越强。由表3中数据可知,在Ni、Mo单掺杂以及Ni-Mo共掺杂之后,Sn-O键的重叠布居数均小于未掺杂时的Sn-O键重叠布居数,说明共价性减弱,离子性增强,导电性增强。Ni-Mo共掺杂之后,Ni-O键的重叠布居数数值更小,表明电子转移加剧,导电性能进一步提高。

图2 能带结构
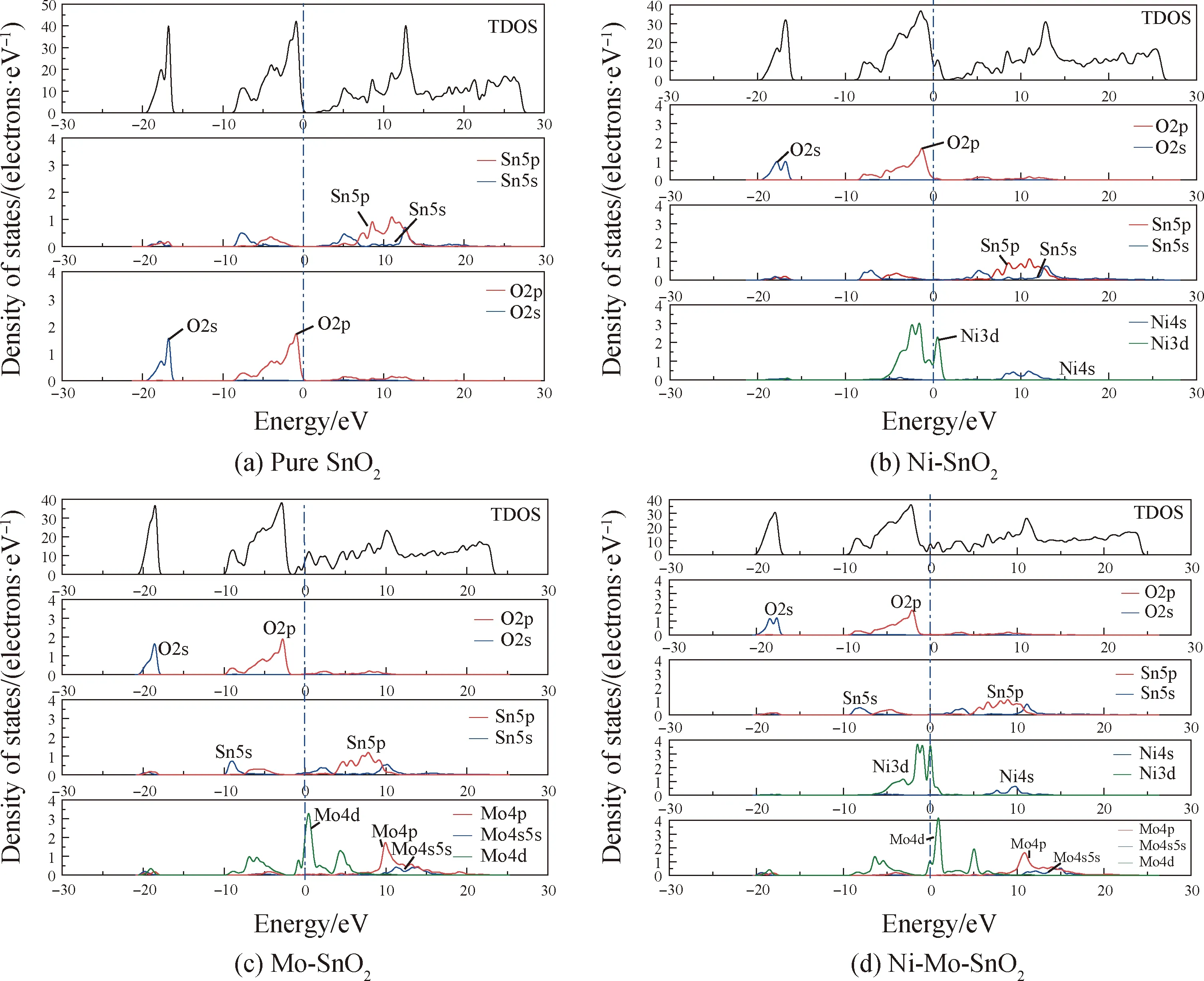
图3 态密度图

表3 原子电荷布居数以及键重叠布居数
2.4 弹性常数分析
弹性常数是分析材料机械性能的重要物理量。根据弹性常数可以得到杨氏模量、体积模量、剪切模量、泊松比等弹性参数,进而分析材料的硬度、脆性、韧性等机械性能。SnO2是四方相金红石结构,属四方晶系,有对称性,四方晶系有6个独立的弹性系数C11、C33、C44、C66、C12和C13,计算结果如表4所示。

表4 各模型弹性系数
对于四方晶系,力学稳定性判断标准为[21]:
C11>0,C33>0,C44>0,C66>0,(C11-C12)>0 (C11+C33-2C13)>0,[2(C11-C12)+C33+4C13]>0
(3)
将表中数据代入上式,计算结果表明各掺杂体系均满足力学稳定性判断标准。对于体积模量、剪切模量而言,常常有两种计算模型,即Voigt模型和Reuss模型,两种模型下四方晶系的剪切模量G、体积模量B与晶体弹性系数Cij之间的关系如下[22-23]:
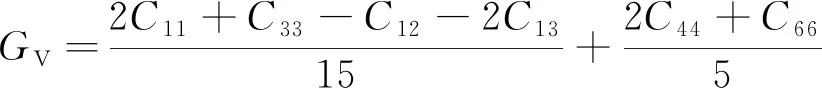
(4)

(5)
式中下标V、R表示Voigt模型和Reuss近似算法。Hill认为这两种算法计算得到的剪切模量G和体积模量B分别为晶体弹性模量的最大值和最小值,而其算数平均值与晶体实际的弹性模量更为接近,Hill模型计算公式如下[24]:

(6)
杨氏模量E的Hill模型计算公式如下:
(7)
为了定量研究单晶的各向异性,Ranganathan等引用了适用于所有晶体的普适弹性各向异性指数AU,计算公式如下[25]:
(8)
由Pugh和Teter的线性关系式,Chen等提出了计算多晶材料硬度的理论公式[26]:
HV=2(k2GH)0.585-3
(9)
式中k=GH/BH。表5所示为通过公式(4)~(9)所计算出的未掺杂SnO2以及掺杂SnO2的体积模量B、剪切模量G、杨氏模量E和硬度HV等参数。

表5 各模型体积模量、剪切模量、杨氏模量、硬度及普适弹性各向异性指数
体积模量B是表征晶体在外力作用下抗体积变化能力的量度。剪切模量G是表征材料抗切应变能力的量度。由表中数据可知,未掺杂SnO2的体积模量和剪切模量要比Mo单掺杂和Ni-Mo共掺杂SnO2的大,因此在外力一定的情况下,未掺杂SnO2的抗体积变化能力要强。由Pugh准则[27],当GH/BH<0.57时,材料表现为韧性,而GH/BH>0.57时,材料表现为脆性。由表中数据可知,Mo单掺杂以及Ni-Mo共掺杂均属于韧性材料,且共掺杂时表现的韧性更好,材料的延展性得到了一定的提高。杨氏模量E定义为线性应力和线性应变的比,其值越小则表明材料的韧性越好,由表可知,Ni-Mo共掺杂时杨氏模量最低,韧性最好,与GH/BH值大小的分析相对应。由表中的硬度值可知,Mo单掺杂后的硬度较之未掺杂SnO2的硬度值有所上升,Ni-Mo共掺杂的硬度值最小,因此共掺杂使得SnO2的硬度降低,有利于后续的加工,且从理论分析上认为,材料的抗熔焊性也得到了提高。普适弹性各向异性指数AU是判断材料是否容易产生裂纹的重要指标,由表中数据可知,掺杂后SnO2的AU值均比未掺杂SnO2的AU值要小,表明SnO2裂纹产生的情况得到了改善,且Mo单掺杂时的数值最小,Ni-Mo共掺杂的数值仅略大于Mo单掺杂。
3 结 论
本文通过Material Studio软件中的CASTEP模块对未掺杂SnO2、Ni单掺杂、Mo单掺杂以及Ni-Mo共掺杂SnO2进行了电性能、力学性能的计算,得到结论如下:
(1)由三种掺杂模型的形成能可知,三种掺杂模型均能稳定存在,且Mo单掺杂的形成能最小,Ni-Mo共掺杂次之。
(2)由三种掺杂模型的能带结构及态密度可知,Ni-Mo共掺杂时带隙值最小,载流子跃迁所需的能量最小,更有利于载流子跃迁,导电性能得到较好改善。
(3)由电荷布居数可知,Ni-Mo共掺杂之后,Ni-O键的重叠布居数数值最小,离子性增强,进而提高了导电性能。
(4)由硬度值可知,Ni-Mo共掺杂时的硬度最小,韧性最强,更有利于AgSnO2的后续加工,便于触头材料成型,且产生裂纹的概率最小。
综合考虑掺杂对于SnO2电性能、力学性能以及热力学性能的影响,Ni-Mo共掺杂可以更有效地改善AgSnO2触头材料的各项性能,对于制备高性能的触头材料提供了理论指导。
猜你喜欢
高中数理化(2022年16期)2022-09-14
高中数理化(2022年4期)2022-03-14
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
原子与分子物理学报(2020年3期)2020-05-15
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
山东化工(2019年1期)2019-01-24
