
增溶策略促非洛地平口服吸收的作用研究
2021-05-08 08:31汪文蝶郑宇钊周建平殷婷婕
中国药科大学学报 2021年2期
李 强,汪文蝶,贾 悦,郑宇钊,周建平,殷婷婕
(中国药科大学药学院药剂系,南京210009)
非洛地平(felodipine)是由瑞典Astra公司开发的二氢吡啶类钙离子通道阻断剂[1],普通片于1988年在丹麦首先上市,现已在多个国家用于高血压的治疗[2]。目前,非洛地平上市产品均为口服制剂,据国家食品药品监督管理局信息,国内市场上,取得生产批文的普通片有7家,缓释片5家,缓释胶囊1家,控释制剂1家。非洛地平属于BCSⅡ(低溶解性、高渗透性)类药物[3],其降压作用呈剂量依赖性,与血药浓度呈正相关[4]。其存在首过效应,生物利用度约20%,主要经肝脏代谢肾脏排泄,血浆蛋白结合率约99%[5]。
对于口服制剂而言,溶出度、渗透性和肠吸收机制是决定其生物利用度的关键因素[6]。非洛地平是BCSⅡ类药物的典型代表,其生物利用度严重受限于在胃肠道中的溶出度。故非洛地平在胃肠道的溶出和吸收特征直接影响其体内的药物疗效。
本研究通过粉末直压法将3种不同粒径原料药压制成非洛地平普通片,原料药粒径分别为200、150、25µm,并通过纳米混悬技术进一步减小粒径,制备非洛地平纳米混悬剂,通过增大溶出比表面积以提高溶出速率;以及通过增溶经典策略——固体分散技术,通过使药物处于高度分散状态提高溶出比表面积、同时使药物以分子或极细胶体、微晶的状态存在以提高非洛地平的溶解度,最终造成溶出速率显著增大。另外,本研究对比上述不同制剂在大鼠十二指肠、空肠、回肠、结肠4个肠段的吸收特征,考察非洛地平溶出度的提高对于其在肠道的吸收速率、表观渗透系数、肠道累积吸收量的影响,进而评价增溶策略对非洛地平口服生物利用度的促进机制。
1材料
1.1 药品与试剂
非洛地平原料药(98%纯度,美国Ark Pharm公司);乳糖(Super T-ab 11SD,荷兰DFEPharma公司);微晶纤维素(安徽山河药用辅料有限公司);硬脂酸镁(湖州展望药业有限公司);乌拉坦(国药集团化学试剂有限公司);吐温80(北京益利精细化工有限公司);交联聚维酮、泊洛沙姆188(德国BASF公司);乙腈、甲醇(色谱纯,安徽天地高纯溶剂有限公司);其他试剂均为市售分析纯。
1.2仪器
RC806D溶出试验仪(广州航信科学仪器有限公司);DP30A单冲压片机(北京国药龙立科技有限公司);Agilent1100高效液相色谱仪(安捷伦科技有限公司);TU-1810紫外可见分光光度计(北京普析通用仪器有限责任公司);RF-5301PC荧光分光光度计(日本岛津公司);Malvern Zetasizer Nano ZSE纳米粒度电位仪(上海思百吉仪器有限公司);DSC200F3差示扫描量热仪(德国耐驰公司)。
1.3 统计学处理
采用GraphPad Prism 8.0分析处理数据,数据统计结果为xˉ±s,组间比较采用t检验。P<0.05认为具有统计学意义。
1.4动物
SPF级SD大鼠,雌性,体重(200±20)g,由南京市青龙山动物繁殖场提供,合格证号:SCXK(苏)2019-0002。所有动物实验均符合动物伦理委员会标准。
2方法
2.1 非洛地平原料药pH-溶解度测定
高效液相色谱条件:Inertsil ODS-SP C18柱(250 mm×4.6 mm,5µm),流动相甲醇-乙腈-水(50∶15∶35),检测波长238 nm,流量1.0 mL/min,柱温30℃,进样量20µL,保留时间26 min。
取过量非洛地平原料药于pH 1.2、2.2、4.5、5.5、6.8、7.4的纯水中,37℃振荡24 h,离心取上清液,高效液相色谱测定。
取过量非洛地平原料药于pH 1.2、2.2、4.5、5.5、6.8、7.4的含1.0%十二烷基硫酸钠(SDS)的溶液中,37℃振荡24 h,离心取上清液,高效液相色谱测定。
2.2 非洛地平普通片的制备
2.2.1 非洛地平普通片制备方法 取粒径分别为200、150、25µm的非洛地平原料药10 mg与乳糖118 mg、微晶纤维素70 mg等量递增预混,过80目筛网3次,加硬脂酸镁2 mg混匀,压片,片重约200 mg,硬度为30~35 N。
2.2.2 非洛地平普通片含量测定 将非洛地平普通片6片研细,精密称取200 mg(约含药10 mg),甲醇溶解,超声,高效液相色谱测定。按外标法计算非洛地平含量和标示量。
2.2.3 非洛地平普通片体外溶出度测定 参照日本药典,选取1.0%SDS增溶,以满足漏槽条件。照溶出度与释放度测定法(《中华人民共和国药典》2015年版四部0931第二法),1.0%SDS 1 000 mL为溶出介质,50 r/min转速,于5,10,15,20,30,45,60,120,180,240,300,420 min,取适量溶液,高效液相色谱法进行测定。
2.3 非洛地平固体分散体
2.3.1 非洛地平固体分散体制备方法 按质量比1∶4∶5称取非洛地平原料药、吐温80、交联聚维酮PVPP,非洛地平原料药、吐温80置梨形瓶,加无水乙醇,超声溶解至澄清,加交联聚维酮,磁力搅拌均匀,45℃减压旋蒸挥发溶剂,真空干燥24 h[7]。
2.3.2 非洛地平固体分散体饱和溶解度测定取过量非洛地平固体分散体于纯水中,37℃振荡24 h,离心取上清液,紫外分光光度法于363 nm处测定吸光度。
2.3.3 非洛地平固体分散体含量测定 非洛地平固体分散体研细,精密称取适量(约含药1 mg),无水乙醇溶解,超声,紫外分光光度法于363 nm处测定吸光度。按外标法计算非洛地平含量。
2.3.4 非洛地平固体分散体差示扫描量热法(DSC)鉴定 分别将非洛地平原料药、吐温80-PVPP、非洛地平与吐温80-PVPP物理混合物、非洛地平固体分散体在速度10℃/min,温度40~180℃下扫描,记录曲线。
2.3.5 非洛地平固体分散体体外释放测定 精密称取适量研细过筛的25µm粒径的非洛地平固体分散体(约含药10 mg)和25µm粒径的非洛地平原料药10 mg,照溶出度与释放度测定法(《中华人民共和国药典》2015年版四部0931第二法),纯水1 000 mL为溶出介质,50 r/min转速,于5,10,15,30,45,60,90,120 min,取适量溶液,高效液相色谱法进行测定。
2.4 非洛地平纳米混悬剂
2.4.1 非洛地平纳米混悬剂制备方法 精密称取非洛地平原料药溶于二氯甲烷,配制50 mg/mL的有机溶液;精密称取泊洛沙姆188溶于纯水,配制0.1%的水溶液。有机溶液与水溶液体积比1∶5,水溶液冰浴5 min,1 000 r/min的转速下,有机溶液快速注入水溶液中,搅拌5 min,探头超声10 min,室温搅拌12 h挥发二氯甲烷[8]。
2.4.2 非洛地平纳米混悬剂粒径与稳定性测定测量非洛地平纳米混悬剂粒径并于常温下放置,观察粒径变化。
按文献配制Krebs-Ringer′s(K-R)液[9],观察KR液稀释非洛地平纳米混悬剂100倍时粒径变化。
2.4.3 非洛地平纳米混悬剂包封率和载药量测定 非洛地平纳米混悬剂于3 000 r/min转速离心10 min,取上清液,甲醇混匀,水浴超声15 min,紫外分光光度法于363 nm处测定吸光度,经外标法计算总含药量。
非洛地平纳米混悬剂于3 000 r/min转速离心10 min,取上清液,4℃下15 000 r/min转速离心30 min,取上清液,甲醇混匀,紫外分光光度法于363 nm处测定吸光度,经外标法计算游离药量。包封率(%)=(总含药量-游离药量)/投药量×100,载药量(%)=(总含药量-游离药量)/(投药量+载体质量)×100。
2.4.4 非洛地平纳米混悬剂体外释放测定 精密量取适量非洛地平纳米混悬剂(约含药10 mg)和25µm粒径的非洛地平原料药10 mg,照溶出度与释放度测定法(《中华人民共和国药典》2015年版四部0931第二法),0.1%SDS 1 000 mL为溶出介质,50 r/min转速,依法操作,于5,10,15,30,45,60,90,120 min,取适量溶液,高效液相色谱法进行测定。
2.5 非洛地平在体单向肠灌流考察
2.5.1 溶液配制 供试混悬液:按上述非洛地平普通片处方,分别精密称取适量200、150、25µm粒径的非洛地平原料药和辅料,K-R液配制成80µg/mL的混悬剂。
供试固体分散体:精密称取适量上述非洛地平固体分散体,K-R液配制成80µg/mL混悬剂。供试纳米混悬剂:精密量取适量上述非洛地平纳米混悬剂,K-R液稀释至80µg/mL。
2.5.2 检测条件与方法 荧光分光光度计于激发波长386 nm,发射波长438 nm处测定非洛地平荧光强度,但非洛地平荧光响应低,测得荧光强度低,定量困难,故选用吐温80增加非洛地平对荧光强度敏感性[10],即非洛地平溶液10 mL中含2%吐温80 1.5 mL。
2.5.3 在体单向肠灌流模型 禁食过夜的雌性SD大鼠(自由饮水),腹腔注射20%乌拉坦(200 mg/kg)麻醉。沿腹中线打开腹腔,选取10~15 cm十二指肠、空肠、回肠、结肠,于两端切口,插管并结扎,用生理盐水洗净肠内容物,再用供试液以2.0 mL/min的流速平衡孵育10 min。伤口用浸有生理盐水的脱脂棉覆盖保湿。入口处用供试液以0.2 mL/min的流速灌流,出液口用已知质量的接收液小瓶收集,每隔30分钟更换下一组供试液和接收液小瓶,实验持续180 min。实验结束后沿插管两端剪下肠段,平放舒展后测量长度和内径。
2.5.4 数据处理 采用重量法矫正灌流液体积,计算药物吸收速率常数(Ka)和表观渗透系数(Papp)[11],由灌流前后药量差值求出每小时单位面积肠壁上累积吸收量,由4个肠段吸收量之和求出累积吸收总量。
2.6 非洛地平药代动力学考察
2.6.1 溶液的配制 非洛地平混悬液:按上述非洛地平普通片处方,分别精密称取适量200,150,25µm粒径的非洛地平原料药及辅料,0.5%羧甲基纤维素钠溶液配制成4 mg/mL的混悬液。
非洛地平固体分散体溶液:精密称取适量上述非洛地平固体分散体,纯水配制成4 mg/mL的固体分散体溶液。
非洛地平纳米混悬液:精密量取适量上述非洛地平纳米混悬剂,纯水稀释至4 mg/mL。
2.6.2 方法学特异性 非洛地平激发波长和发射波长分别为386和438 nm;空白血浆激发波长为291 nm,发射波长为352 nm;吐温80无荧光吸收。因此,血浆、吐温80对非洛地平测定不干扰。
2.6.3 给药方案及血样采集 将实验前禁食12 h(自由饮水)的雌性SD大鼠随机分成5组,分别为200µm非洛地平原料药与辅料组、150µm非洛地平原料药与辅料组、25µm非洛地平原料药与辅料组、非洛地平固体分散体组、非洛地平纳米混悬液组。80 mg/kg剂量灌胃,给药后0.25,0.5,0.75,1,1.5,2,3,4,6,8,10,24 h于大鼠眼眶后静脉丛取血0.5 mL,置预先肝素化的EP管中,10 000 r/min离心5 min,分离上层血浆,-20℃储存。
2.6.4 样品与数据处理 非洛地平血浆样品室温解冻,吸取170µL,加2%吐温80 30µL,荧光分光光度计测得荧光强度。采用PK Solver按非房室模型处理数据,计算主要药代动力学参数。
3 结果与讨论
3.1 非洛地平原料药pH-溶解度测定
如图1-A所示,非洛地平在不同pH的溶液中溶解度极低,达不到漏槽条件;图1-B显示,1.0%SDS显著提高非洛地平溶解度,满足漏槽条件,非洛地平溶解度无pH依赖性。

Figure 1 pH-solubility curves of felodipineactive pharmaceutical ingredient(API)A:Felodipine APIin the water;B:Felodipine APIin the 1.0%SDSsolution
3.2 非洛地平普通片
3.2.1 药物含量测定 结果如表1所示,药物含量均在标示量的95.0%~105.0%范围内,符合规定。
3.2.2 溶出度 图2表明,不同粒径原料药的非洛地平普通片均在7 h内释放量达85.0%以上,符合规定。且原料药粒径越小,溶出速率和程度越高。

Table1 Determination of felodipinecontent(xˉ±s,n=3)

Figure 2 Dissolution curve of felodipine tablets based on 200,150,25µm particle sizes of bulk drug in 1.0%SDSsolution(xˉ±s,n=3)*P<0.05 vs150µm tablets group
3.3 非洛地平固体分散体
3.3.1 药物饱和溶解度 非洛地平固体分散体及其原料药饱和溶解度分别为(179.03±5.47)µg/mL和(2.80±0.12)µg/mL。该结果表明固体分散体显著提高非洛地平在水性介质的饱和溶解度。
3.3.2 药物含量测定 该固体分散体中主药含量为(9.88±0.28)%。
3.3.3 DSC鉴定 由图3可知,非洛地平在147.49℃出现吸热峰,吐温80-PVPP载体无吸热峰,非洛地平和载体的物理混合物只出现非洛地平吸热峰,说明非洛地平与载体之间没有相互作用或相互作用较弱,但固体分散体吸热峰消失,表明固体分散体中非洛地平以无定形或分子形式存在。
3.3.4 体外释放 图4表明,非洛地平固体分散体在1 h内累积释放量为(86.69±2.54)%,原料药累积释放量为(12.24±2.31)%,可见,将非洛地平制备成固体分散体后,其体外释放显著提高。
3.4 非洛地平纳米混悬剂
3.4.1 粒径与稳定性 如图5-A所示,非洛地平纳米混悬剂粒径为(168.90±6.22)nm,PDI:0.11±0.06,粒径分布较均一。如图5-B所示,非洛地平纳米混悬剂常温下48 h内粒径稳定。图5-C表明,K-R液稀释100倍的非洛地平纳米混悬剂在12 h内亦能保持粒径稳定。
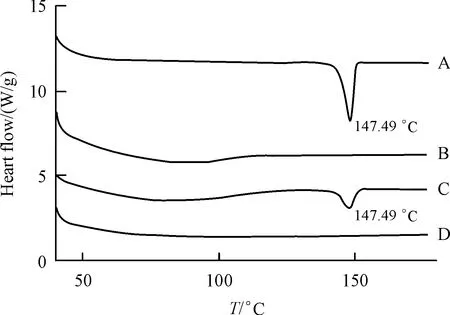
Figure 3 DSCthermograms of felodipine API,Tween 80-PVPP,phys⁃ical mixture and felodipine solid dispensionA:Felodipine API;B:Tween 80-PVPP;C:Physical mixture;D:Felo⁃dipinesolid dispension
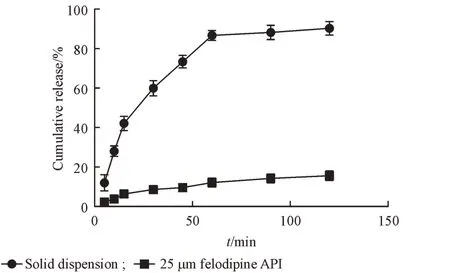
Figure 4 In vitro release curves of felodipine solid dispension and 25µmfelodipine APIin water(xˉ±s,n=3)
3.4.2 包封率和载药量 非洛地平纳米混悬剂包封率为(78.98±2.31)%,载药量为(39.46±1.24)%。
3.4.3 体外释放 图6所示,非洛地平纳米混悬剂在90 min内累积释放量为(87.52±5.48)%,原料药累积释放量为(20.39±1.03)%,因此,纳米混悬剂可显著提高原料药的体外释放。
3.5 非洛地平在体单向肠灌流考察
大鼠在体单向肠灌流法测得指标与人体吸收指标有较好的相关性,该法建立模型所测的数据能应用于人体吸收的预测[12],已成为美国食品药品管理局认可的研究药物吸收的模型之一[13],故本研究采用此模型来研究非洛地平的胃肠道吸收机制。由于胃肠道中存在的P-糖蛋白(P-gp)可影响药物在肠道吸收效果,而非洛地平不是P-gp底物,故本研究中不考虑P-gp的作用。
3.5.1 不同制剂在十二指肠、空肠、回肠、结肠中的吸收速率 如图7-A和7-B所示,同一肠段(结肠除外)中,非洛地平的Ka随原料药粒径减小而增大,非洛地平固体分散体、纳米混悬剂较普通片均可进一步提高Ka(P<0.05)。以上结果说明,增大以非洛地平为代表的BCSⅡ药物的溶出速率,可有效提高药物在肠道的吸收速率。
图7-C数据显示,同一非洛地平制剂在十二指肠、空肠、回肠的Ka均优于结肠(P<0.05),但十二指肠、空肠、回肠之间的Ka无显著性差异(P>0.05),说明非洛地平主要吸收部位在小肠,且在小肠无特定吸收肠段,而大肠吸收速率低。结肠可能因吸收效果不佳,不同制剂之间Ka无显著性差异(P>0.05)。

Figure 5 Particle size figures of felodipine nanosuspension(xˉ±s,n=3)A:Felodipine nanosuspension at 0 h;B:Felodipinenanosuspension at 48 h;C:Felodipine nanosuspension diluted in K-Rsolution at 12 h

Figure 6 In vitro release curves of felodipine nanosuspension and 25µm felodipine APIin 0.1%SDSsolution(xˉ±s,n=3)
3.5.2 不同制剂在十二指肠、空肠、回肠、结肠中的表观渗透系数 药物在大鼠体内Papp大于2.0×10-5cm/s,表现为高渗透性[14]。如图8所示,不同制剂不同肠段的非洛地平Papp均大于2.0×10-5cm/s,因此非洛地平属于高渗透性药物,限制其肠道吸收的主要因素是低溶解性。
图8-A数据表明,粒径减小,普通片的Papp在全肠段均有所增加;图8-B表明,相比于最低粒径的普通片,固体分散体和纳米混悬剂的Papp进一步增大;分析图8-C数据可知,非洛地平在所有制剂中均在十二指肠的Papp最大,且显著高于结肠(P<0.05)。可能因普通片溶解度低、渗透性高,溶出药物几乎都渗入肠壁,导致同一粒径的非洛地平普通片在不同肠段Papp无明显差异(P>0.05)。综上,对于BCSⅡ药物,提高其胃肠道内的溶出速率,有增大其Papp的潜力。

Figure 8 Apparent permeability coefficient of felodipine tablets,solid dispersion and nanosuspension in the duodenum,jejunum,ileum and colon(xˉ±s,n=3)A:200µm,150µm and 25µm felodipine tablets;B:25µm felodipine tablets,felodipine solid dispersion and felodipine nanosuspension;C:Felodip⁃inetablets,solid dispersion and nanosuspension*P<0.05 vs150µmtabletsgroup;#P<0.05 vs25µmtabletsgroup;△P<0.05 vs colon group
3.5.3 不同制剂在十二指肠、空肠、回肠、结肠中的累积吸收量与累积吸收总量 与上述Ka和Papp的结果趋势相一致,如图9所示,在普通片中原料药粒径越小,非洛地平的累积吸收量越大;通过新技术进一步提高非洛地平溶解度和溶出速率,可较低粒径普通片再度提高其在各肠段的相应吸收量(P<0.05)。最后,非洛地平累积吸收总量也呈相同趋势:普通片,粒径越小,累积吸收总量越大;固体分散体和纳米混悬剂累积吸收总量最大,进一步证明增加以非洛地平为代表的BCSⅡ类药物的溶解度和溶出速率,可显著促吸收(P<0.05)。

Figure 9 Absorption quality and total absorption quality of felodipine tablets,solid dispersion and nanosuspension in the duodenum,jejunum,ileum and colon(xˉ±s,n=3)A:Absorption quality of 200µm,150µm and 25µm felodipine tablets;B:Absorption quality of 25µm felodipine tablets,solid dispersion and nano⁃suspension;C:Total absorption quality of felodipine tablets,solid dispersion and nanosuspension*P<0.05 vs150µm tablets group;#P<0.05 vs25µm tablets group
3.6 药代动力学考察
如表2和图10所示,非洛地平固体分散体cmax、AUC和F最高,非洛地平纳米混悬剂次之,最大粒径非洛地平普通片最小。在普通片中,粒径减小非洛地平cmax、AUC和F依次递增。结果证明,增加以非洛地平为代表的BCSⅡ类溶解度和溶出速率,口服后能提高胃肠道吸收量,从而增加在体内的有效浓度,进而提高口服生物利用度。
4结 论
本研究制备了原料药粒径为200µm、150µm、25µm的非洛地平普通片,通过制剂增溶新技术制备了非洛地平纳米混悬剂和固体分散体,研究增溶策略对以非洛地平为代表的BCSⅡ类药物口服吸收的影响。pH-溶解度曲线验证非洛地平溶解性差,大鼠在体肠灌流实验结果表明非洛地平渗透性高,因此限制非洛地平等BCSⅡ类药物口服吸收的关键因素其是低溶解性;增加以非洛地平为代表的BCSⅡ类药的饱和溶解度和溶出速率,能显著提高其在胃肠道吸收速率和累积吸收量,从而最终提高生物利用度。

Table2 Pharmacokinetic parametersof felodipinetablets,solid dispersion and nanosuspension(xˉ±s,n=5)

Figure 10 Plasma concentration-time curves of felodipine tablets,solid dispersion and nanosuspension(xˉ±s,n=5)
猜你喜欢
中国经济周刊(2021年22期)2021-12-07
中华养生保健(2020年4期)2020-11-16
环球时报(2020-02-20)2020-02-20
传媒评论(2019年6期)2019-10-14
中国经济信息(2017年17期)2017-09-09
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中西医结合心血管病电子杂志(2016年13期)2016-11-30
中国实用医药(2016年16期)2016-07-26
中学生数理化·中考版(2015年11期)2015-09-10
