
DNA环境中硒代胸腺嘧啶和腺嘌呤碱基对的激发态性质和光物理机理的理论研究
2021-07-11 16:14方业广张腾烁崔刚龙方维海
高等学校化学学报 2021年7期
彭 沁,方业广,张腾烁,崔刚龙,方维海
(北京师范大学化学学院,理论及计算光化学教育部重点实验室,北京100875)
天然碱基腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶和尿嘧啶是脱氧核糖核酸(DNA)和核糖核酸(RNA)的基本组成单元部分,对核酸序列识别、遗传信息存储、DNA聚合酶复制等均有重要作用.它们具有许多优异的物理化学性质[1],如光稳定性,可防止紫外线照射下的生物光损伤.为了揭示其光保护的分子机制,在过去十几年中,大量的实验和理论研究探索了在不同环境,如真空、溶液、DNA或RNA中,碱基及碱基对的激发态性质和动力学行为[2,3].现在,普遍认为这些碱基的光稳定性归因于它们可以通过锥形交叉结构,从激发态超快地衰减到基态,最终将能量耗散到环境中[4~10].
与天然碱基不同,将碱基上的氧原子进行硫取代生成的硫代碱基具有明显不同的激发态和光物理性质[11,12].在过去几年中,实验和理论研究探索了硫代碱基的激发态性质和衰减动力学等[13~23].Harada等[24]发现4-硫代胸腺嘧啶三重态量子产率接近100%;随后的瞬态吸收实验表明,4-硫代胸腺嘧啶在水溶液中存在皮秒时间尺度的系间窜跃过程[25].Crespo-Hernández等[26,27]发现4-硫代胸腺嘧啶在离子溶液中存在超快的系间窜跃动力学,持续时间小于240 fs,解释了较弱的室温磷光现象.他们也研究了在不同溶液中2-硫代胸腺嘧啶和2-硫代尿嘧啶亚皮秒的系间窜跃动力学,认为S1暗态在激发态弛豫过程中起着重要作用[28].郑旭明课题组[29]利用共振拉曼技术探索了2-硫代尿嘧啶在S2态上的驰豫动力学.最近,Crespo-Hernández等[30~33]进一步研究了2,4-二硫代胸腺嘧啶,发现其接近100%的三重态量子产率和超快的系间窜跃动力学.不难发现,天然碱基是以超快的内转换过程为主,但是硫代碱基则是以系间窜跃过程为主要的激发态弛豫通道.为了理解其奇特的实验现象背后的物理本质,一些理论课题组利用高精度的电子结构和非绝热动力学方法系统地研究了它们的激发态和光物理性质[34~47].
由于氧、硫和硒属于同一族元素,硒代碱基在过去几年也引起了一些实验方面的关注[48~55],但主要集中在电子的基态.Crespo-Hernández等【56】研究了6-硒代鸟嘌呤(6SeG)在溶液中的激发态性质,发现了单重态到三重态有超快的系间窜跃和超高的三重态量子产率;但是,相比于硫代体系,三重态的寿命较短.此外,Huang课题组[51,57]成功合成了含硒代碱基的核酸,发现在DNA中4-硒代胸腺嘧啶和6-硒代鸟嘌呤的吸收光谱发生明显的红移.此外,他们在RNA中也发现2-硒代尿嘧啶的吸收光谱会发生明显的红移[55].
在计算方面,Russo等[58,59]用含时密度泛函理论(TD-DFT)方法探索了硒代胸腺嘧啶和鸟嘌呤的光物理性质.Manae和Hazra[60]使用多态完全活化空间二级微扰理论(MS-CASPT2)方法计算了硫和硒代胸腺嘧啶的吸收光谱.Mai等[61]使用MS-CASPT2和二阶代数图表构造法ADC(2),探索了2-硒代尿嘧啶(2SeU)的激发态弛豫路径,并开展了非绝热动力学模拟.他们认为与2-硫代尿嘧啶相比,2SeU的三重态寿命减少了3个数量级,主要原因是缺少第二个三重态的参与、自旋-轨道耦合的增强以及T1/S0势能面交叉结构和T1极小能量结构的较小能量差.最近,本课题组[62]使用高精度MS-CASPT2方法,研究了6SeG的激发态弛豫路径,发现增强的T1/S0旋-轨耦合导致了较短的三重态寿命.随后继续使用MS-CASPT2方法,分别研究了1个和2个硒原子取代的2SeU,4SeU和24SeU的激发态性质和光物理机理,发现不同位点的硒取代会显著影响垂直和绝热激发能、激发态性质和弛豫路径[63].
DNA环境非常复杂,如氢键作用可能对碱基的光物理性质产生较大的影响,但至今大多数关于硒代碱基的理论计算研究并没有较为合理地考虑DNA环境的影响.为了考虑DNA环境中互补碱基对的氢键对碱基的光物理机理的影响,本文使用量子力学/分子力学组合方法和高精度的电子结构方法,即QM(MS-CASPT2//CASSCF)/MM方法,研究和比较了DNA环境中2-硒代胸腺嘧啶和腺嘌呤碱基对(2SeT-A)以及4-硒代胸腺嘧啶和腺嘌呤碱基对(4SeT-A)的激发态性质和光物理过程.
1 计算方法
1.1 计算模型
根据实验研究提供的结构信息,利用AMBER16程序包中的NAB模块建立了DNA序列为5′-CTTCTTGTCCG-3′∶3′-GAAGAACAGGC-5′的双链结构[64],并放置于80 nm×80 nm×80 nm的立方体水盒子中.然后,加入20个钠离子,使得体系变为电中性.在建立的初始体系基础上,在前1000步中,整个DNA序列保持冻结,优化水分子及钠离子;在剩下的2000步里对整个体系能量进行最小化处理,不施加任何限制.然后加热20 ps,使体系从0 K升温至300 K;最后,进行1 ns的恒温和恒压的分子动力学模拟.在分子力场的结构优化和动力学模拟中,DNA结构和钠离子使用Amber ff99sb力场描述[65],而水分子使用TIP3P模型描述[66].随后,在分子动力学模拟得到的稳定结构基础上,将其中一个单链的第五个胸腺嘧啶碱基的2和4号位置的氧原子分别替换为硒原子生成含有2-硒和4-硒代胸腺嘧啶碱基(2SeT或4SeT)的DNA双链结构,其中与硒取代胸腺嘧啶碱基配对的是互补链的腺嘌呤碱基A.
1.2 QM/MM计算
所有电子结构计算均在量子力学/分子力学(QM/MM)方法框架下进行[67~70].QM区域包含2SeT或4SeT及与之配对A碱基(2SeT-A或4SeT-A).MM区域则由剩余的DNA结构及所有的离子和水分子构成.对于QM/MM极小能量结构和交叉点结构的优化,QM区域的电子结构方法选用完全活化空间自洽场(CASSCF)方法[71].在CASSCF计算中,单重态和三重态均采用5态平均计算,每个电子态的权重为0.2.在单重态计算中平均了S0~S4这5个电子态,而在三重态中平均了T1~T5这5个电子态.由于该方法不能充分考虑动态电子的相关效应,因此,在QM/MM单点能量计算的时候,QM区域选用多态完全活化空间自洽场二级微扰理论(MS-CASPT2)方法[72].同样地,在MS-CASPT2计算中均采用5态平均,态平均计算参数与CASSCF计算一致.同时,采用0.2原子单位的虚能级移动来避免入侵组态问题[73].根据本课题组之前的经验和测试及近期Zobel等[74]的测试结果,电离和亲核势(IPEA)校正设置为零[75].同时使用Cholesky分解技术快速而准确地计算双电子积分[76],截断阈值设置为10-4.在CASSCF和MS-CASPT2计算中,活化空间选用14电子11轨道(本文支持信息中图S1和图S2).C,H,O和N原子选用cc-pVDZ基组,重原子Se则选用较大的aug-cc-pVDZ基组[77,78].旋-轨耦合常数利用平均场近似方案计算[79~81],表示为
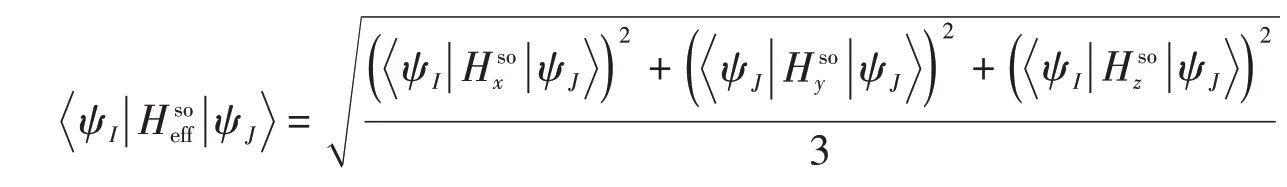
式中:ψI和ψJ是单重态和三重态的电子波函数;Hsox,Hsoy和Hsoz是旋-轨算符的x,y和z分量.所有的QM/MM计算使用与TINKER6.3.2软件包连接的MOLCAS8.0软件包.
2 结果与讨论
2.1 基态的稳定结构和光谱性质
采用QM(CASSCF)/MM方法首先优化了2SeT-A和4SeT-A在S0态上的极小能量结构(图1,S0-MIN).尽管这些结构都具有类似平面的对称性,但是也存在一些明显的结构差异.当O原子变为Se原子时,键长明显变长,如2SeT-A中的C2―Se7键长伸长到0.184 nm.此外,在2SeT-A和4SeT-A的S0-MIN结构中,与C―O和C―Se键直接相连的化学键的长度也会发生明显变化.
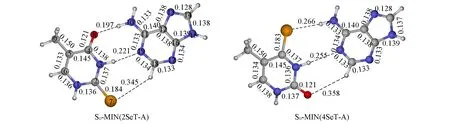
Fig.1 QM(CASSCF)/MMoptimized minima of 2SeT-A and 4SeT-A base pair in the S0 states
Franck-Condon区域的激发态性质对于理解2SeT-A和4SeT-A的激发态弛豫动力学具有重要作用.在优化得到的S0极小能量结构处,QM(MS-CASPT2)/MM计算表明2SeT-A和4SeT-A的S1态是具有1nπ*性质的暗态,而S2态是具有1ππ*性质的明态.但是,它们的1nπ*和1ππ*态的电子组态涉及的前线轨道不同.对于2SeT-A,1nπ*态对应Se7原子的n电子激发到嘧啶和C2-Se7双键π*轨道.相比之下,4SeT-A的1nπ*态激发Se8原子的n电子到嘧啶和C4-Se8双键的π*轨道(图2).这些差异导致2SeT-A和4SeT-A的1nπ*态的垂直激发能不同,其中2SeT-A为336.7 kJ/mol,而4SeT-A降低到243.1 kJ/mol(表1).有关1ππ*态,π*轨道与1nπ*态中的相同,而π轨道不同.2SeT-A中的π轨道主要由Se7原子的p轨道组成;而4SeT-A中是由Se8原子的p轨道组成(图2).同样地,1ππ*态的垂直激发能也略有不同,其中2SeT-A为378.2 kJ/mol,而4SeT-A为347.3 kJ/mol(表1).在2SeT-A和4SeT-A中,具有1ππ*性质的更高的电子激发单重态能量约为460.0 kJ/mol,分别比最低的1ππ*态高约84.0和113.0 kJ/mol,实验所用激发波长难以到达.因此,本文主要研究从最低的1ππ*态出发的激发态弛豫机理.
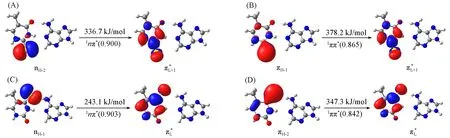
Fig.2 Molecular orbitals relevant to the lowest two excited singlet states at the Franck-Condon points of 2SeT-A and 4SeT-A

Table 1 QM(MS-CASPT2)/MM calculated vertical excitation energies(kJ/mol)at the S0 minima of 2SeT-A and 4SeT-A
2.2 激发态极小能量结构
除了S0态的稳定结构,也用QM(CASSCF)/MM方法优化了S2,S1,T2和T1激发态的极小能量结构.对于2SeT-A,分别优化出了C―Se键沿着嘧啶环平面向上翘(-U)和向下翘(-D)两套激发态结构(本文支持信息图S3).因为这两套激发态极小能量结构对应的激发态能量比较接近,因此文中主要讨论其中C―Se键沿着嘧啶环平面向上翘的结构(加个后缀-U).图3和图4分别为2SeT-A-U的激发态结构示意图及S2,S1,T2和T1激发态极小能量结构相对于S0稳定结构键长的变化值.可以发现,这些结构的变化主要位于2SeT和4SeT部分,而A碱基的贡献几乎可以忽略不计.这些几何结构特征和上述讨论的激发态电子结构特征是一致的.与S0态的稳定结构相比,S2态的C2―Se7键长变化最大,伸长了0.039 nm;其次是C6―N1,N1―C2,N3―C4和C4―C5键长,分别变化了约0.002,0.001,-0.001和0.001 nm.在S1,T2和T1态的极小能量结构中,也是C2―Se7键长变化最明显,分别变化了0.023,0.025和0.031 nm,略小于S2态的C2―Se7键长;但是C6―N1,N1―C2,C2―N3,N3―C4和C4―C5键长变化相对较大(图4).QM(MS-CASPT2)/MM计算表明,2SeT-A-U和2SeT-A-D的S2,S1,T2和T1的绝热激发能分别为313.8和312.5、257.3和268.2、257.7和254.4、242.7和256.5 kJ/mol.2SeT-A-D的激发态极小能量结构及对应键长的变化见图S4和图S5(本文支持信息).
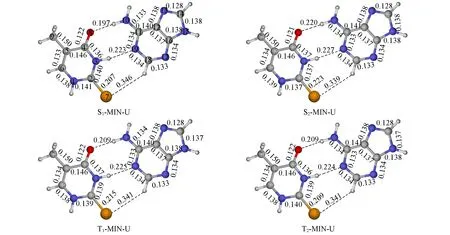
Fig.3 QM(CASSCF)/MMoptimized minima of 2SeT-A-U base pair in the S1,S2,T1 and T2 states

Fig.4 Bond-length changes of QM(CASSCF)/MM optimized excited-state minimum-energy structures of 2SeT-A-U relative to corresponding ground-state structures
对于4SeT-A,除了U和D两套激发态极小能量结构外,还优化得到了C―Se键与嘧啶环基本共平面的结构(-P).同样地,这3套结构对应的能量均比较接近(本文支持信息图S13),因此主要讨论其中C―Se键沿着嘧啶环平面向上翘的结构(加个后缀-U).图5和图6分别为4SeT-A-U的激发态平衡结构及S2,S1,T2和T1相关激发态的平衡结构相对于S0平衡结构对应键长的变化值.同样可以发现,这些激发态结构的变化主要发生于4SeT,而A碱基部分的结构仍然变化较小.与2SeT-A-U的激发态极小能量结构不同,4SeT-A-U相对于S0稳定结构变化最大的是C4―Se8键.其中,S2态的极小能量结构的C4―Se8键长变化最大,增加了0.042 nm;其次为C4―C5,C5―C6和C6―N1键长,分别变化了-0.002,0.001和-0.001 nm.在S1,T2和T1态的结构中,C4―Se8键长也是变化最大的,分别为0.015,0.021和0.018 nm,但是小于S2态的C4―Se8键长变化;而N1―C2,C2―N3,C3―N4和C6―N1键长相对变化也较大(图6).对于4SeT-A-U(4SeT-A-D,4SeT-A-P),QM(MS-CASPT2)/MM计算的S2,S1,T2和T1态的绝热激发能分别为299.6(289.1,297.1),210.5(211.3,212.1),204.2(216.7,206.3)和199.6(208.8,209.6)kJ/mol.对应的4SeT-A-D和4SeT-A-P的激发态结构及相关键长变化见图S6~图S9(本文支持信息).
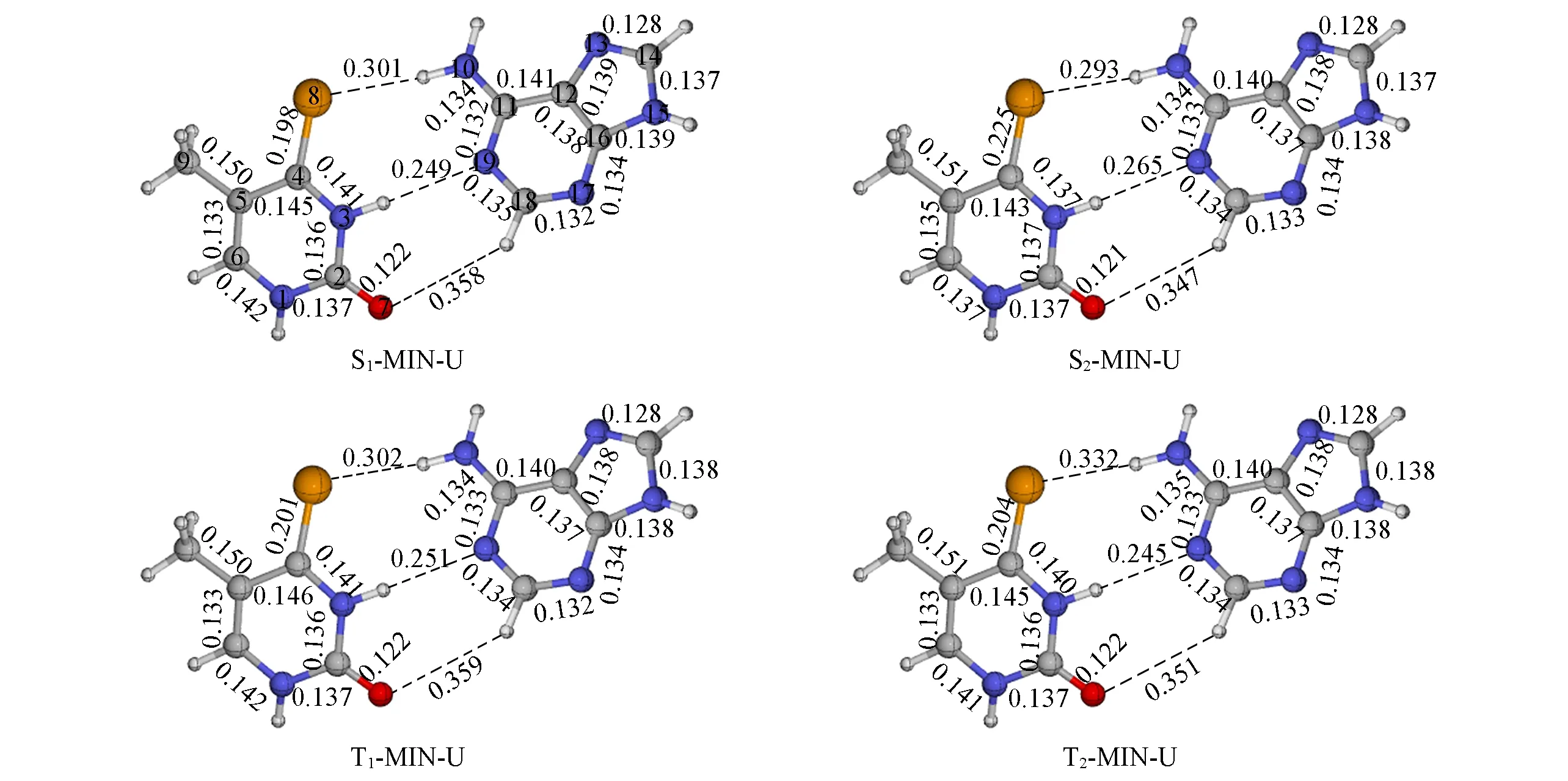
Fig.5 QM(CASSCF)/MMoptimized minima of 4SeT-A-U base pair in the S1,S2,T1 and T2 states
Fig.6 Bond-length changes of QM(CASSCF)/MM optimized excited-state minimum-energy structures of 4SeT-A-U relative to corresponding ground-state structures
2.3 交叉点结构
众所周知,不同势能面之间的交叉结构对于激发态弛豫过程有着重要作用,是连接不同势能面之间的“桥梁”.对于2SeT-A,在S2-MIN-U和S2-MIN-D的极小能量结构处,S2和S1的能量已经比较接近,QM(MS-CASPT2)/MM计算相对能量分别为313.8/296.2和312.5/297.9 kJ/mol.因此,在这两个极小能量结构附近,处于S2态的体系可以有效地衰减到S1态.此外,还优化得到了T1/S0的交叉结构T1S0(图7).在该结构处,C2―Se7键长被拉长到0.246 nm,Se7-C2-N1-N3二面角变为125.1°.在QM(MS-CASPT2)/MM计算级别,T1和S0的能量为265.3和252.3 kJ/mol.对于4SeT-A,找到了S2/S1和T1/S0的两个交叉结构S2S1和T1S0(图7).它们的C4―Se8键分别拉长到0.278和0.247 nm,而Se8-C6-C2-N1二面角分别为175.8°和146.9°.在S2S1结构,QM(MS-CASPT2)/MM计算的S2和S1态的能量分别为374.5和359.8 kJ/mol,而在T1S0结构中,计算的T1和S0能量分别为228.4和214.6 kJ/mol.
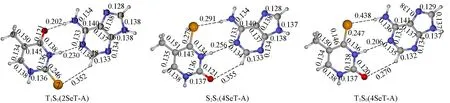
Fig.7 QM(CASSCF)/MMoptimized intersection structures of 2SeT-A and 4SeT-A
2.4 激发态的失活机理
根据优化得到的极小能量结构和交叉结构,并结合计算得到的线性内插内坐标(LIIC)路径,分析得到了2SeT-A和4SeT-A在DNA环境中的主要激发态失活机理.对于2SeT-A体系,当光照到达Franck-Condon区域的S2态(FC S2)后,存在2条不同的弛豫路径,分别到达S2势能面中的两个极小能量结构S2-MIN-U和S2-MIN-D.对于路径Ⅰ,如图8所示,体系首先弛豫到S2-MIN-U.在该结构处,由于S2和S1态的能量比较接近,能量差仅为17.6 kJ/mol;因此,可以通过内转换有效地弛豫到S1态,然后弛豫到S1-MIN-U,该过程没有能垒.在S1态上面,由于S1,T2和T1态具有较小的能差,因此S1态到T2和T1态的系间窜跃过程是允许发生的.但是,较大的S1/T1旋-轨耦合(395.0 cm-1)使得S1→T1的系间窜跃过程比较有利.到达T1-MIN-U后,2SeT-A可以通过T1/S0交叉点衰减到S0态,该过程要克服22.6 kJ/mol的能垒.在该结构处,较大的T1/S0旋-轨耦合(432.6 cm-1)将有助于T1→S0的系间窜跃过程,但是较大的能垒使T1态有可测量的寿命.如图S10(本文支持信息)所示,路径Ⅱ与路径Ⅰ类似.体系首先无能垒弛豫到S2-MIN-D,然后继续衰减到S1-MIN-D,最后到T1-MIN-D.不同之处在于,在路径Ⅱ中,从T1-MIN-D到T1/S0交叉点需要克服73.6 kJ/mol的能垒,使T1态的寿命更长.
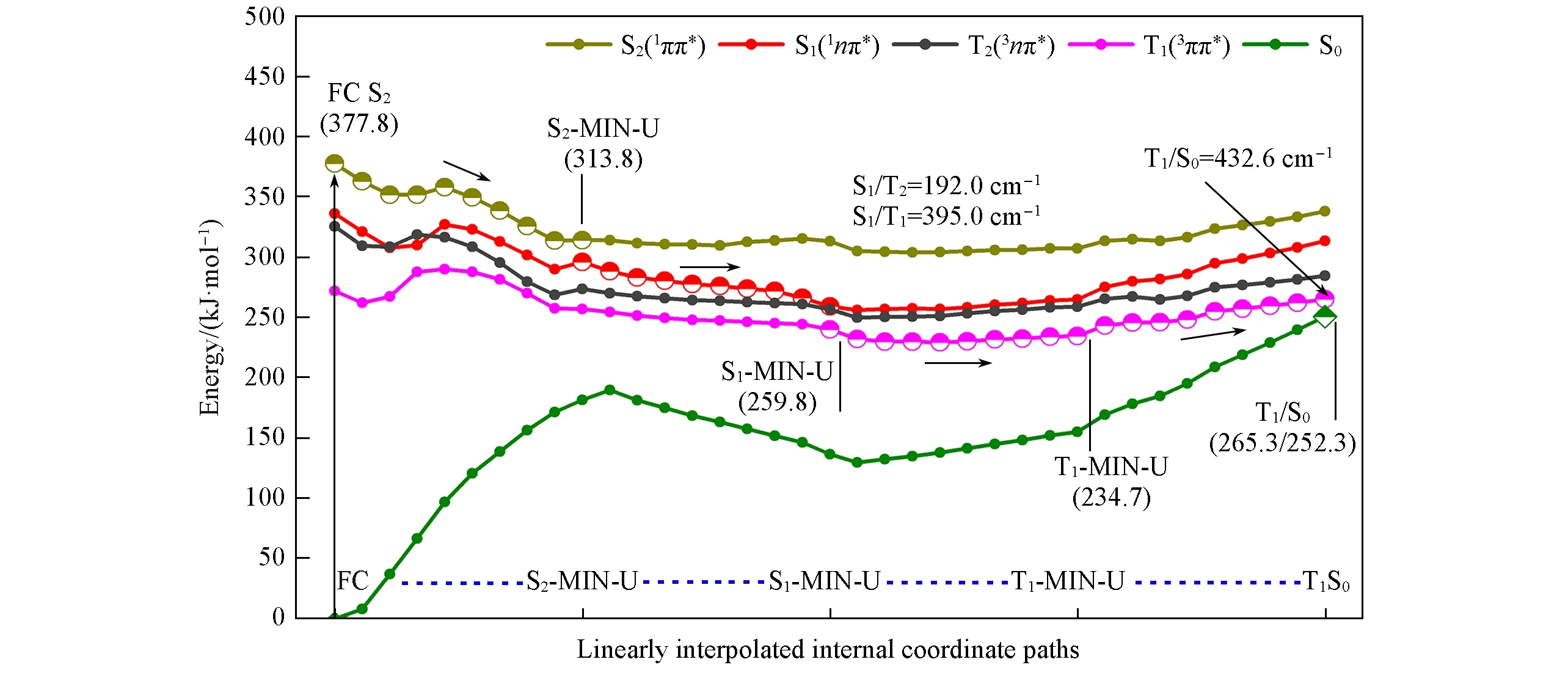
Fig.8 Main excited relaxation path of 2SeT-A in DNA calculated by QM(MS-CASPT2)/MM(the conformation of C─Se bond up along the pyrimidine ring plane)
对于4SeT-A,光激发到S2态Franck-Condon区域后,可以有3个不同的路径.对于路径Ⅰ,分子首先弛豫到S2-MIN-U,但是从S2-MIN-U到S2/S1圆锥交叉点需克服60.7 kJ/mol的能量.在S1态上面,存在大量S1,T2和T1态近简并的区域,如图9所示.沿着这些路径,具有较大的S1/T1旋-轨耦合(410.0 cm-1)的S1→T1系间窜跃过程更为有利;而S1→T2系间窜跃过程,由于较小的S1/T2旋-轨耦合(39.1 cm-1)则相对较慢.这些现象符合EI-Sayed规则[82].因为S1和T2态具有相同的nπ*电子结构,而S1和T1态则具有不同的nπ*和ππ*电子结构.因此,在S1→T1无辐射跃迁过程中,总的角动量可以守恒,有利于系间窜跃发生.到达T1-MIN-U后,考虑到T1/S0交叉结构处的较大旋-轨耦合(373.0 cm-1),4SeT-A可以进一步衰减到S0态,但是该过程要克服28.9 kJ/mol的能垒.因此,体系会在T1态上停留一段时间.路径Ⅱ和Ⅲ(本文支持信息图S11和图S12)与路径Ⅰ的激发态弛豫过程接近,因此,3条路径都有可能对4SeT-A在DNA环境中的激发态失活做出贡献,具体的重要性要进一步的非绝热动力学模拟才能完全确定.

Fig.9 Main excited relaxation path of 2SeT-A in DNA calculated by QM(MS-CASPT2)/MM(the conformation of C—Se bond up along the pyrimidine ring plane)
3 结 论
使用QM(MS-CASPT2//CASSCF)/MM方法,系统研究了在DNA环境中2-硒和4-硒代胸腺嘧啶和腺嘌呤碱基对2SeT-A和4SeT-A的激发态性质和光物理过程.尽管2SeT-A和4SeT-A具有相似的S2(ππ*),S1(nπ*),T2(nπ*)和T1(ππ*)电子激发态,但是由于涉及不同的n和π轨道,它们的激发态行为存在较大差异,导致2SeT-A的垂直和绝热激发能都比4SeT-A高.此外,无论2SeT-A还是4SeT-A在DNA环境中均存在多个具有不同构象的激发态极小能量结构和激发态失活途径.尽管这些激发态弛豫路径有相似之处,基本是按照S2→S1→T1的光物理过程失活到最低的三重T1激发态,使T1态具有可测量的寿命.但是具体的S2→S1内转换过程在2SeT-A和4SeT-A中还是有显著区别.前者基本是个没有能垒的过程,后者则需要克服60.7 kJ/mol的能垒.总体上可以看出,2SeT-A和4SeT-A在DNA环境中的激发态弛豫路径主要是系间窜跃到最低三重态.计算工作也表明不同位置的硒取代胸腺嘧啶在DNA环境中具有显著不同的激发态性质和光物理机理,这些差异将有助于理解硒取代碱基激发态性质和光物理机理的复杂环境效应.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210117.
猜你喜欢
汕头大学学报(自然科学版)(2020年4期)2020-12-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
天然产物研究与开发(2018年5期)2018-06-13
中国有色金属学报(2018年2期)2018-03-26
中成药(2016年8期)2016-05-17
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
中学化学(2015年8期)2015-12-29
原子与分子物理学报(2015年3期)2015-11-24
化工进展(2015年3期)2015-11-11
读写算·教研版(2014年12期)2014-09-01
