
笼状团簇Au6B8N24的第一性原理研究
2021-08-05 06:01屈文欢郭静静刘蓬勃赵荟艳温美沙
河北北方学院学报(自然科学版) 2021年7期
屈文欢,郭静静,刘蓬勃,赵荟艳,温美沙
(1.河北北方学院 理学院,河北 张家口 075000;2.河北师范大学 物理学院,河北 石家庄 050026)
0 引 言
近年来越来越多的学者寻找稳定的团簇作为纳米结构材料和电子器件。高度对称集群笼形结构表现出的不寻常稳定性和反应性激发了物理学家和化学家极大的兴趣[4-7],从碳原子出发除了构建平面石墨和金刚石结构还可以凝结成非常稳定的笼状结构,例如C60、C70和C20[8-9]。
与此同时,近十年来金纳米结构也引起实验和理论研究者的广泛关注,人们对含金团簇的兴趣越来越大。金纳米团簇也由于在生物、催化和纳米技术等领域的广泛应用,在近二十年来引起了人们的广泛关注。考虑到Au元素在元素周期表中的位置,它是一种不寻常的相对论性元素,这种相对论效应使得很多Au团簇和纳米管成为可能,例如Au32、Au72和Au42等[1-6]。除此之外,它的成键特征使得Au元素表现出亲水性,近一步增强了Au-Au之间的相互作用。
人们发现掺杂不同的元素后团簇分子的稳定性会得到相应的提高,由于过渡金属元素的存在,这类团簇分子呈现出了各种各样新的物理和化学性质。在众多过渡金属元素中,Au元素因为独特的物理和化学性质被人们广泛应用于多面体团簇结构的构建中。因为B元素、N元素与C元素在元素周期表上相邻,它们的化学性质有相似之处。考虑到将Au元素、B元素和N元素结合起来可能构建出稳定的团簇结构。本文提出了一个稳定的金属笼状团簇结构Au6B8N24,并对其进行优化,计算和分析它的稳定性和电子结构。
1 理论基础与计算方法
1.1 密度泛函理论
密度泛函理论起源于Thomas-Fermi模型,是以Hohenberg-Kohn定理和Kohn-Sham提出的“外势场是电荷密度的单值函数”为可靠的理论依据[10-11],自1970年,人们开始在研究分子和固体电子模型中广泛应用密度泛函理论。
交换关联泛函在密度泛函理论中占有重要的地位。Hobenberg-Kohn方程在形式上将多电子基态特性问题转换成了有效的单电子问题。然而只有将有相互作用的粒子的全部复杂性归入到交换关联相互作用泛函中才可以更加简单、严密地导出单电子方程。交换关联泛函包括以下几种形式:局域密度近似(LDA)[12]、广义梯度近似(GGA)[13]、完全非局域密度泛函、杂化密度泛函等。
1.2 计算软件
Material Studio(MS)计算软件采用量子力学(QM)、线性标度量子力学(linear scaling QM)、分子力学(MM)、分子动力学(MD)、耗散粒子动力学(DPD)等先进的模拟计算方法和思想,对小分子、晶体、非晶体、团簇以及高分子材料的结构及其性质进行分析研究。MS作为研究密度泛函理论的主要计算工具被广泛地应用于材料科学的研究。MS中包含很多模块,其中的量子力学模块包括CASTEP(平面波赝势方法)、DMol3(原子轨道线性组合方法)、QMERA(量子力学/分子力学杂化方法)、ONETEP(线性标度方法)等。根据本文研究的结构和性质,主要选择使用了CASTEP和DMol3模块。
CASTEP(平面波赝势方法)是利用赝势替代内层电子,基于密度泛函理论,采用平面波函数描述价电子。被广泛用于材料科学、固体物理、化学化工等领域。
DMol3(原子轨道线性组合方法)包含PW91[2]、PNE[14]、BLYP[15]等多种交换关联势,主要采用的是数值函数描述原子轨道,具有较高的数值基组精度。DMol3不仅可以预测材料的光学、电子学、热力学性能,还可以研究气相、溶液、表面及其他固态环境中的化学反应,主要用于晶体材料、非晶体材料、有机分子、团簇等周期及非周期体系研究。
1.3 计算方法
本文是基于自旋极化密度泛函理论(DEF)使用DMol3软件包首先对Au6B8N24团簇结构进行了结构的优化,优化过程没有自旋限制和对称性限制。后采用包含极化函数的双数值缀加基组(DNP)[3],选取广义梯度近似(GGA)和PBE交换关联泛函。考虑到Au的相对论效应,我们对核内电子采用DFT半芯赝势(DSPP)的方法进行处理[1]。为了论证Au6B8N24团簇结构的稳定性,我们进行了振动频率分析和分子动力学模拟,分子动力学模拟中设置参数为总时间5 ps,时间的步长1.0 fs,步骤5 000步。最后利用高斯09软件包对结构进行了自然轨道(NBO)分析[7]。
2 结果与讨论
2.1 结构稳定性
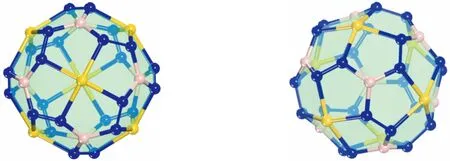
(a)Au6B8N24球棍模型的正视 (b)Au6B8N24球棍模型的侧视
图1中的Au6B8N24结构的球棍模型,黄色球表示Au原子、粉色球表示B原子、蓝色球表示N原子。由图中可以看出,Au6B8N24结构具有O点群对称性,以1个Au原子、3个N原子和1个B原子形成1个五元环,这样的4个五元环以Au原子为中心相对接,一共由这样的24个五元环相互连接堆积而成。Au6B8N24结构还有另外一种理解方式,就是以B原子为中心与3个N原子和1个Au原子形成3个五元环相连接,同样也是由24个相同的五元环链接堆积而成。其中每个五元环中形成1个N-N键,2个同一类型的Au-N键,2个同种类型的B-N键。
表1中给出了Au6B8N24团簇结构使用PBE交换关联泛函的计算结果,其中包括对称性(Sym),原子间平均键长(dN-N、dAu-N、dB-N),能量带隙(Eg),平均每原子结合能(Eb)。显示Au6B8N24团簇结构具有O对称性,N原子间平均键长为1.368Å,Au和N原子间平均键长为2.096Å,B原子和N原子间平均键长为1.483Å。该结构的平均每原子结合能为4.479eV/atom,最高占据轨道和最低占据轨道间的带隙为0.314 eV。
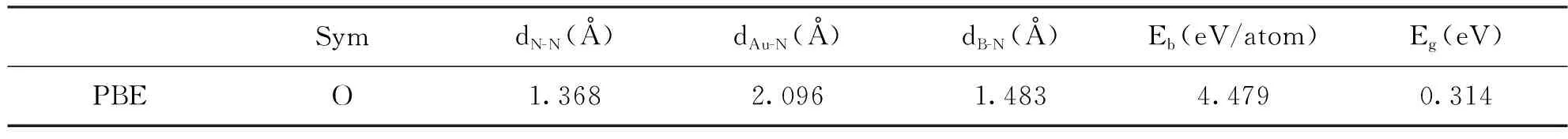
表1 Au6B8N24性质计算结果汇总
对优化后的Au6B8N24结构进行振动频率分析,得到结构的拉曼光谱(图2)。设置入射光为488.0 nm、温度为300 K。可以看出该结构的振动频率在65.1~1 117.7 cm-1之间,表明Au6B8N24结构没有虚频,是稳定的团簇结构。图中对应标注了振动频率峰值的振动模式,在65.4 cm-1处对应的振动模式主要是Au原子的振动模式,在118.6 cm-1处对应的主要是Au原子和N原子的拉伸模式,在432.7 cm-1处主要对应B原子和N原子的振动模式,到最大振动处对应N原子的振动模式。这里的理论计算结果可以为实验的合成提供参考。
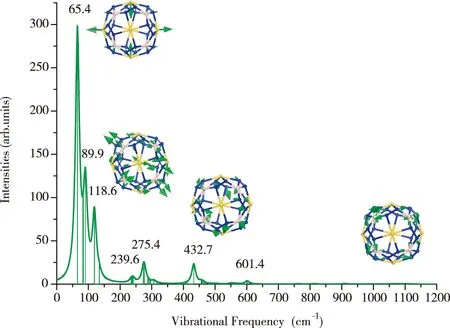
图2 Au6B8N24团簇结构的拉曼光谱
在NVE系综中模拟该结构的分子动力学(MD),从而进一步确认Au6B8N24团簇结构的稳定性。首先设置初始温度为1 600 K,时间步长为1.0 fs,模拟总时间5 ps,分子动力学模拟的总步数设为5 000步。对优化后的Au6B8N24团簇初始结构进行分子动力学模拟,发现整个过程中Au6B8N24团簇结构保持原有的结构。之后分别设置初始温度为2 000 K、2 400 K、2 800 K、3 200 K、3 600 K,时间步长为1.0 fs,模拟总时间5 ps,分子动力学模拟的总步数设为5 000步,进行5次模拟。模拟结果发现随着温度增加结构发生轻微扭曲,但是仍保持良好的原始结构,当初始温度为3 600 K时,Au6B8N24团簇结构发生严重扭曲,化学键发生严重破坏。所以我们又设置初始温度为3 400 K,对应的有效温度为1 557.0 K,再次进行分子动力学模拟,做出温度为3 400 K时分子动力学步数随温度的变化曲线图,如图3(a)所示,以及在1 ps、2 ps、3 ps、4 ps、5 ps时的拓扑结构,如图3(b)所示。可以看出在初始温度为3 400 K,有效温度为1 557.0 K时Au6B8N24团簇结构保持其原有的位形,证明Au6B8N24团簇结构具有良好的动力学稳定性。
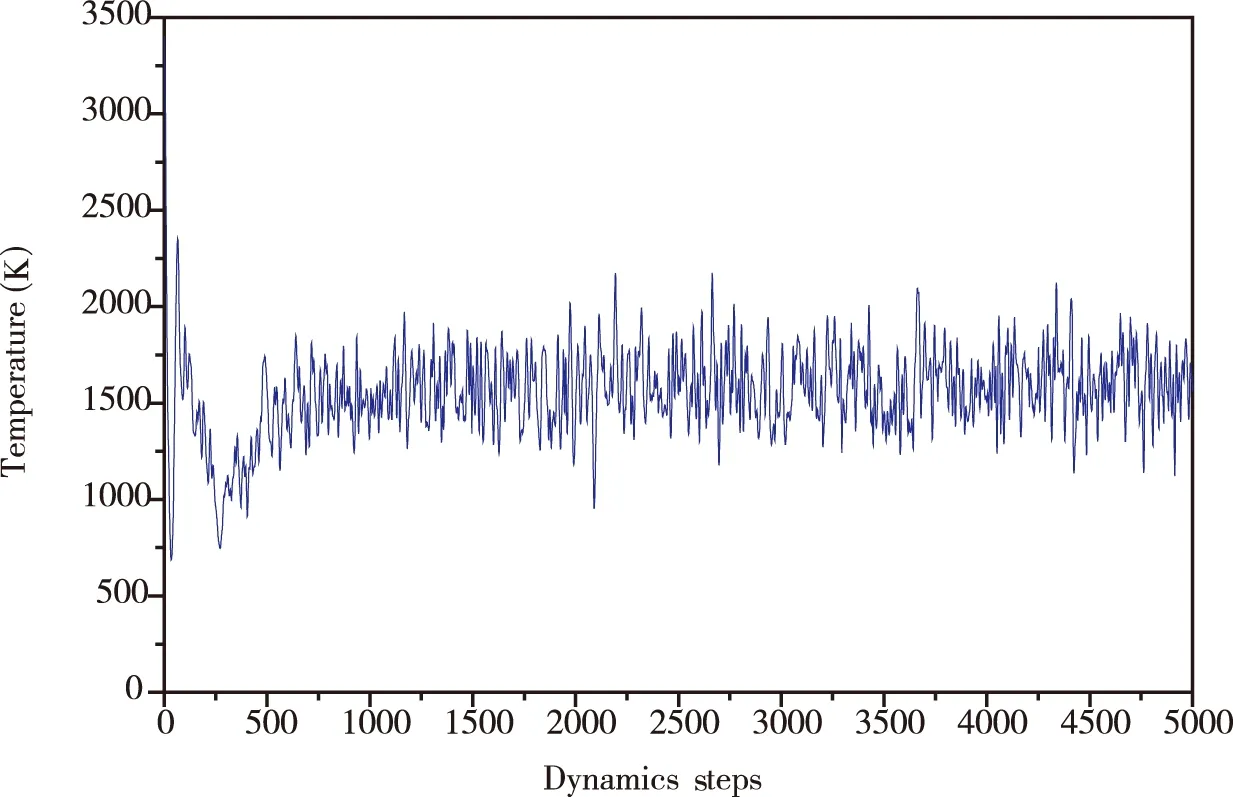
(a)Au6B8N24团簇随温度变化的分子动力学步数,初始温度设置为3 400 K
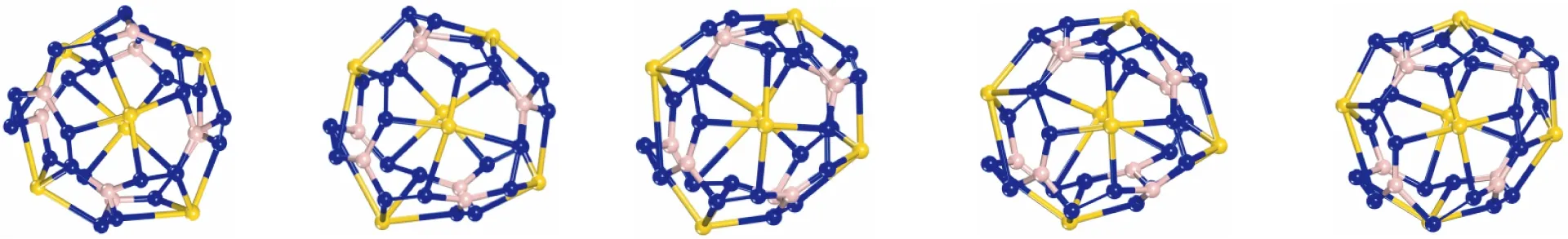
1ps 2 ps 3 ps 4 ps 5 ps
2.2 电子性质
为了进一步说明结构稳定性,我们对Au6B8N24团簇结构的电荷转移进行分析,研究了它的电子性质。表2显示Au原子和N原子失电子,B原子得电子。Au原子大约有0.444 e转移到N原子上,B原子上大约有0.102 e转移到N原子上。
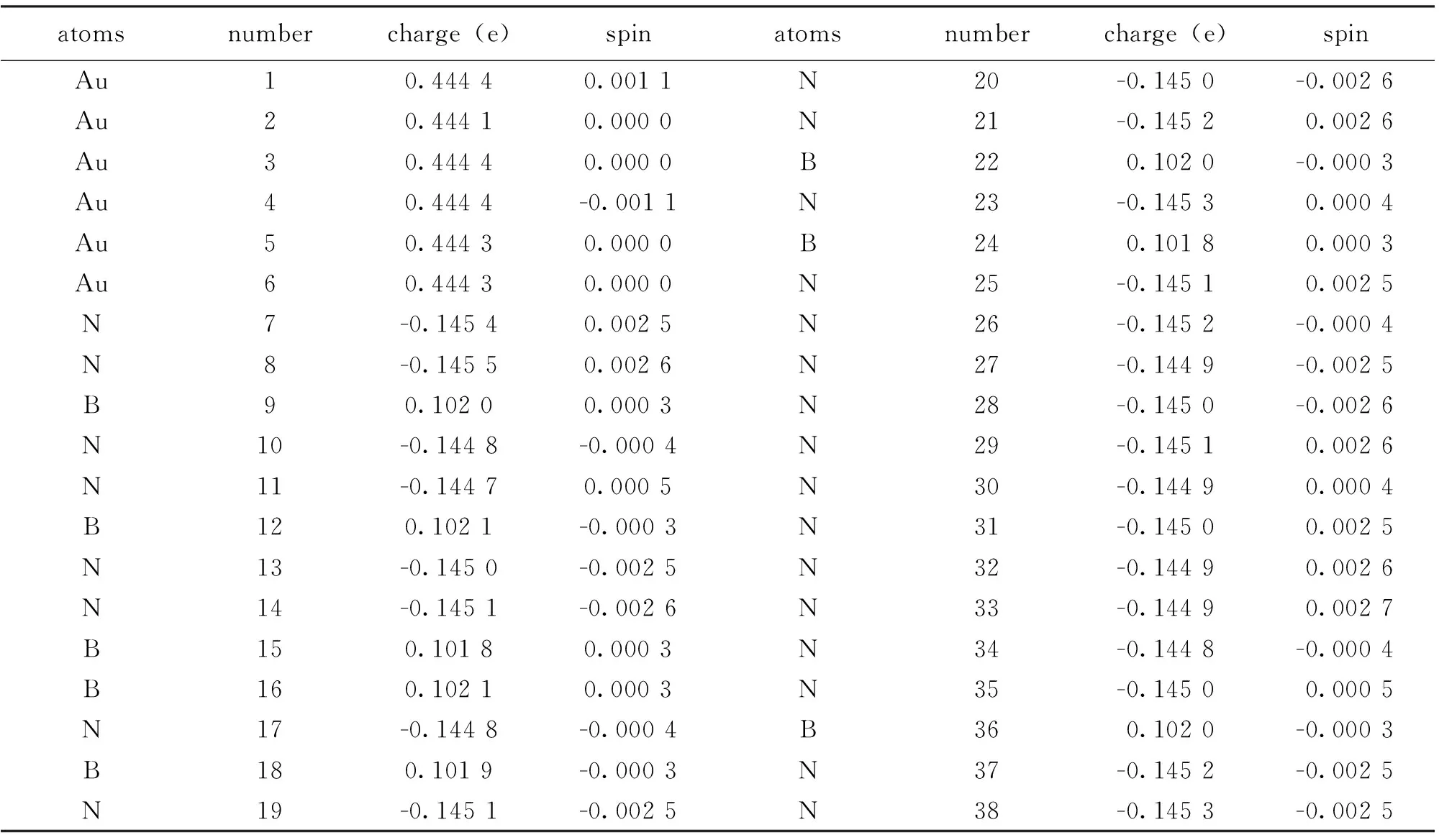
表2 利用Hirshfeld方法对Au6B8N24团簇进行电荷转移分析
图4、图5、图6分别为Au6B8N24结构的分波态密度(PDOS)、最高占有分子轨道图(HOMO)和最低未占分子轨道图(LUMO)。表明在费米面附近主要存在N原子和B原子的p电子贡献,从HOMO和LUMO图中也可以看出Au原子上存在明显的d轨道特征。
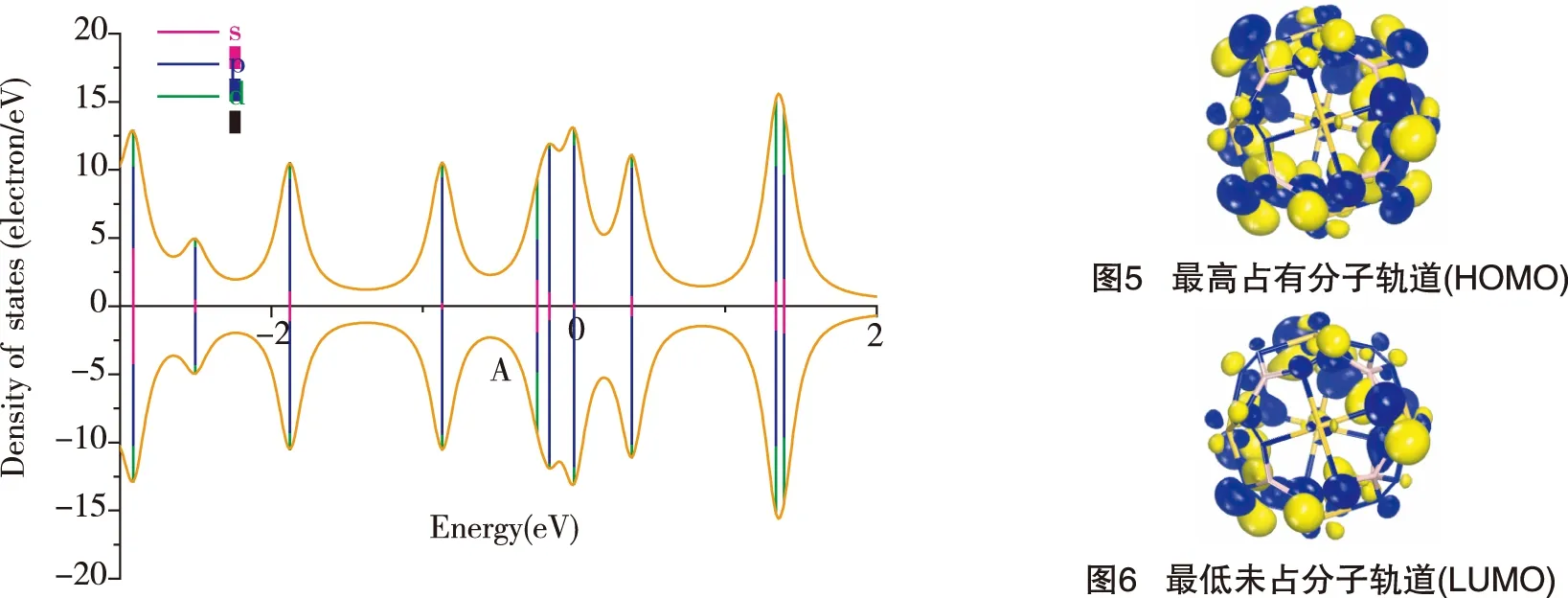
图4 Au6B8N24的分波态密度(PDOS)
图7给出Au6B8N24结构的差分电荷密度图,图中蓝色表示电荷聚集,黄色表示电荷耗散。由图可以明显看出Au原子和B原子上有明显的电荷耗散,其中Au原子上还存在明显的d电子轨道特征,N原子上存在明显的电荷聚集。

图7 Au6B8N24结构的差分电荷密度
对Au6B8N24团簇结构自然成键轨道(NBO)分析表明,图8中(1)~(4)显示Au原子上有4个d孤对电子(LP),占据数为1.97~1.99 |e|。(5)显示N原子的1个孤对电子(LP),占据数为1.81 |e|。(6)、(7)是Au原子和N原子之间形成的σ键,杂化形式分别为Au原子的sp2.25d1.04杂化(23.34% s、52.46% p、24.20% d)和N原子的sp3.39杂化(22.75% s、77.25% p),以及Au原子的sp2.60d1.54杂化(19.44% s、50.54% p、30.03% d)和N原子的sp4.75杂化(17.39% s、82.61% p)。由成键轨道可以看出,电子都向Au原子堆积,这与理论相符。(8)是N原子和N原子之间形成的σ键,由1个N原子的sp2.36杂化(29.78% s、70.22% p)和另1个N原子sp2.39杂化(29.47% s、70.53% p)组成。(9)表示N原子和N原子之间形成的π键是由N原子中的p电子组成。(10)和(11)是B原子和N原子之间形成的σ键,(10)的元素组分是N元素77.82%,B元素22.18%;(11)元素组分是N元素22.86%,B元素77.14%,基本上正好相反。
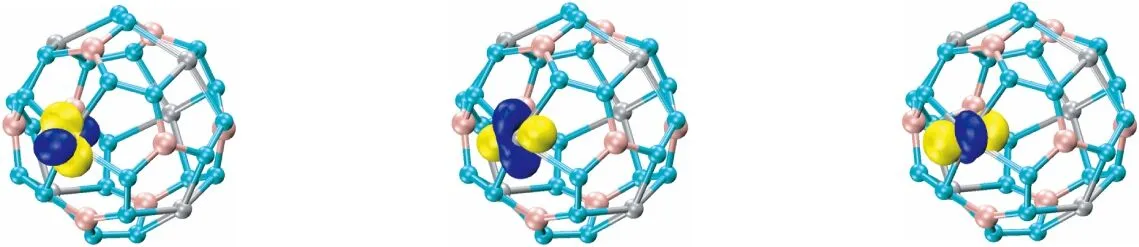
(1) (2) (3)
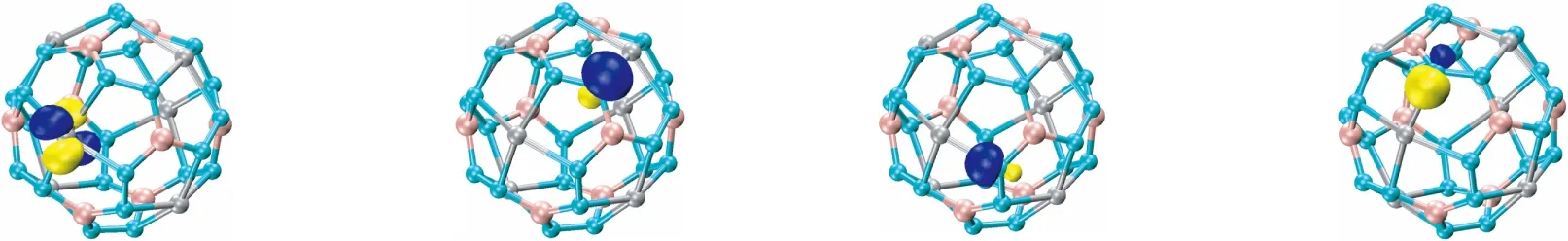
(4) (5) (6) (7)

(8) (9) (10) (11)
3 结论与讨论
1)首先,对Au6B8N24多面体团簇结构进行结构优化,优化后的结构可以看作是以1个Au原子、3个N原子和1个B原子围成1个五元环,4个五元环以Au原子为中心相连接,一共有24个五元环堆集而成。结构中Au原子、N原子和B原子都处于相同的位置。2)其次,为了探究结构的稳定性,分析了结构的振动频率,做出相应的拉曼谱,发现没有虚频,说明了结构的稳定性。并且对结构进行了热力学稳定性分析近一步确定稳定性。设置不同的初始温度,从1 600 K开始依次递增,确定在初始温度为3 400 K,有效温度为1 557.0 K时Au6B8N24团簇结构保持其原有的位形,证明Au6B8N24团簇结构具有良好的动力学稳定性。而后又分析了结构的电子性质,结构中Au原子和N原子失电子,B原子得电子,这与做出的态密度图以及HOMO、LUMO图相符合,从图中可以看出Au原子上存在明显的d轨道特征。3)最后,对结构进行了NBO分析,结果表明Au原子上有4个d孤对电子(LP),占据数为1.97-1.99 |e|;N原子上有1个孤对电子,占据数为1.81|e|;Au原子和N原子之间形成σ键;N原子和N原子之间形成一种σ键和一种π键成键轨道;B原子和N原子之间形成σ键。以上数据为今后的理论和实验研究提供理论依据。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
空气动力学学报(2022年4期)2022-08-23
北京航空航天大学学报(2022年7期)2022-08-06
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
黑龙江大学自然科学学报(2022年1期)2022-03-29
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
读写算·教研版(2016年8期)2016-05-07
中学化学(2014年4期)2014-09-09
