
金刚石(111)/Al 界面形成及性能的第一性原理研究*
2021-10-08 08:56孙士阳迟中波徐平平安泽宇张俊皓谭心任元
物理学报 2021年18期
孙士阳 迟中波 徐平平 安泽宇 张俊皓 谭心 任元
(内蒙古科技大学机械工程学院,包头 014010)
针对金刚石/Al 界面的形成和性能,采用第一性原理计算方法,研究了Al 原子在H 终止金刚石表面的吸附及其迁移行为,以及金刚石/Al 界面的结构与黏附功.结果表明:Al 原子吸附对金刚石原子表面结构不敏感,且表面迁移激活能非常小,其原因是Al 与H 原子间没有形成化学键,仅有少量电荷转移,为物理吸附;由吸附位置生长而形成的金刚石/Al 界面为亚稳结构,不具有能量稳定性.本文结果为理解金属纳米掩膜的形成机理提供重要的理论参考.
1 引 言
金刚石因饱和的原子键和稳定的晶体结构,而具有高硬度、高热导率、宽带隙、极高化学惰性和极好的生物相容性等优异性能,在切削工具、复合材料、电子器件、生物传感等领域得到广泛的应用[1−4].纳米结构的量子效应和非线性[5],进一步扩大了金刚石材料在科学前沿的应用.其中,金刚石纳米线的应用和研究最为广泛[6−8].如何制备出纳米线结构,并加以尺寸控制,是金刚石纳米线研究的基础.
金刚石纳米线的制备方法有多种,其中模板法应用最为广泛.Masuda 等[9]以阳极氧化铝为模板,采用微波等离子体化学气相沉积法制备了规则有序的金刚石纳米线和类金刚石纳米管.Yang 等[10]以纳米金刚石颗粒作为硬掩膜,采用化学气相沉积结合反应离子刻蚀法制备了金刚石纳米线.Okuyama 等[11]报道了一种以二维分散固体SiO2颗粒阵列作为掩膜,在氧等离子体气氛中,用反应离子刻蚀法制备得到周期性排列的金刚石纳米阵列.Smirnov 等[3]开发出更为简便的金属掩膜法,采用Ni 金属膜热处理后的纳米颗粒作为掩膜,再利用氧等离子刻蚀法制备出直径为10—60 nm的金刚石纳米阵列.
上述研究展示出模板法是一种可成功制备出金刚石纳米线的有效方法.纳米线的结构尺寸及成本都取决于模板的制作工艺和材料选取.以金属掩膜法为例,由于缺乏金属纳米颗粒形成的理论支持,需要耗费实验资源去优化工艺参数.因此,对金刚石表面金属膜形成机理及其结构控制的研究势在必行.金刚石表面金属纳米颗粒的形成,涉及到金刚石/金属界面结构和性质,以及金属原子在金刚石表面的吸附、迁移和生长等行为.Scholze等[12]和Stampfl 等[13]分别采用第一性原理方法比较了金刚石(111)表面重构结构的表面能和表面电子结构.Larsson 和Lunell[14]研究了金刚石(111)卤素元素终端的稳定性;文献[15−18]理论计算了金刚石(001)表面的分子吸附及其吸附后表面的电负性;刘峰斌等[19]研究了不同金属与金刚石(100)表面形成界面的模型及其结合性能.
虽然这些研究对了解金刚石表面结构和金刚石/金属界面的稳定结构有很大帮助,但由于研究的侧重点不同,仍无法揭示金属在金刚石表面形成界面的过程,以及形成界面后的结构和性质.
针对金刚石/Al 界面的形成和结构,本文采用第一性原理方法,研究了Al 原子在H 终止金刚石(111)表面的吸附和迁移行为;并在此基础上根据界面黏附功分析金刚石/Al 界面的形成机理.
2 计算模型
计算采用基于密度泛函的第一性原理计算程序软件包(VASP)[20]进行.利用投影增强波(PAW)[21]方法描述离子核与价电子之间的相互作用;利用广义梯度近似中的Perdew-Burke-Ernzerhof[22]方法描述电子之间的交换相关势;利用共轭梯度法优化几何结构[23];并用Helleman-Feymann 力作为收敛判据,其精度设置0.02 eV/Å;利用Methfessel-Paxton 方法[24]计算电子占据率,其中Smearing的展宽优化后确定为0.1 eV;电子自洽迭代矩阵对角化时选用直接求逆的残量最小化算法RMMDIIS 算法[25],电子自洽循环迭代精度设置为1.0 ×10–5eV;平面波截断能选取650 eV;布里渊区的积分采用Monkhorst-Pack 方法[26],优化后的仿真模型选取网格为[7 × 7 × 1].原子迁移采用微动弹性带方法寻找能量最低路径(MEB),在初态和终态间线性插值5 个像点.
金刚石薄膜生长常形成(111)织构,金刚石/Al 界面的形成可认为是由Al 原子在金刚石(111)表面吸附、迁移、生长而形成.其中吸附和迁移是界面形成的基础,很大程度上决定了界面的初始结构.本文建立了金刚石(111)微观表面结构(slab)模型,如图1 所示.金刚石slab 模型包含16 个原子格点,即4 × 4 面心立方原胞;生长方向设置6 层原子,底部3 层固定,其余原子松弛;为避免表面空间对吸附原子的影响,真空层需要足够大,文中设置为40 Å.由于金刚石(111)表面存在悬键,易发生表面重构形成碳的二聚体[27].考虑到沉积态金刚石生长环境,H 终止金刚石(111)面为最稳定结构[28].在(111)表面上,存在4 个高对称吸附位置,分别为Top,Br,T4 和H3 位.其中Top 位表示表面C 原子正上方(垂直于slab),Br 位置在表层相邻C 原子中间正上方;T4 位置为表层C 原子三重洞位,且为亚表层C 原子正上方;H3 位置是表层C 原子另一个三重洞位,且在第4 层C 原子正上方.界面模型由金刚石基底、金属Al 层和真空层组成.其中,金刚石基底(111)表面采用H 终止结构,基底设置12 个原子层,3 层固定其余松弛,每层有2 × 2 原子格点,共40 个C 原子;金属端设置10 层Al 原子,并全部松弛,其位置根据稳定吸附位置考虑;真空层同样设置为40 Å.分子动力学的模型采用H 终止金刚石(111) slab 模型,slab 模型包含16 个原子格点,即4 × 4 面心立方原胞;生长方向设置6 层原子,底部3 层固定,其余原子松弛;表面吸附1 层Al 原子(3 × 3),真空层设置为40 Å.模拟采用宏观正则系综(NVT),初始温度为800 ℃,最终温度为室温,等温区时间步长0.5 fs,步数设置1000 步,退火过程中时间步长1 fs,总步数设置10000 步.
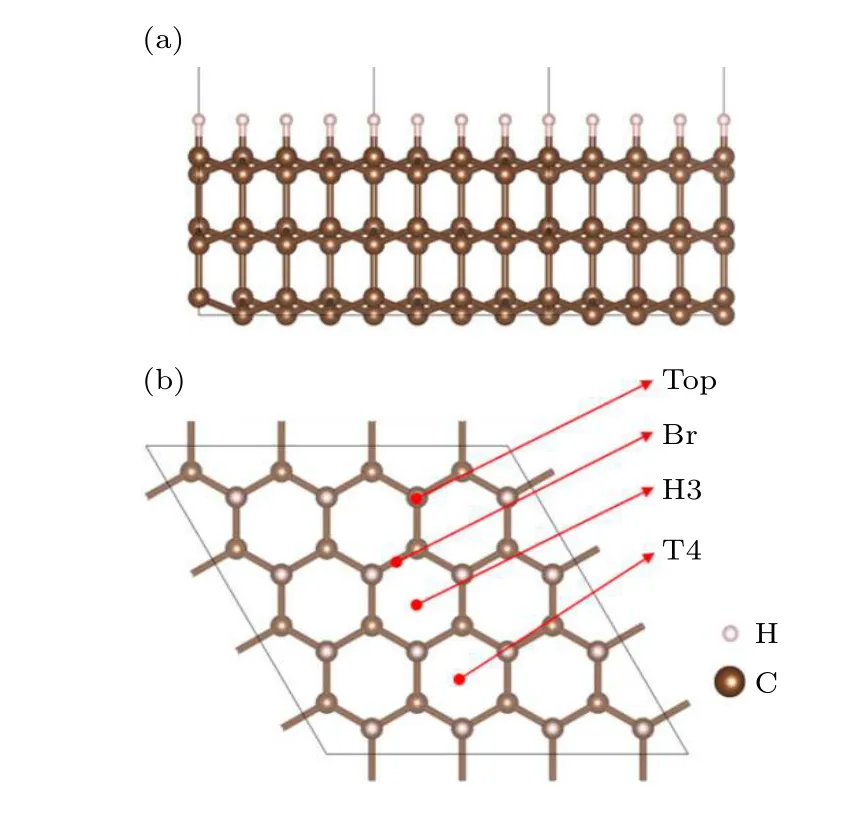
图1 H 终止金刚石(111)表面模型,其中棕色球表示C原子,白色小球表示表面H 原子 (a)模型原子分布的立体结构示意图;(b)金刚石(111)表面格点位置示意图Fig.1.H-Ter diamond(111) surface model:(a) Three-dimensional structure diagram of model atom distribution;(b) location of diamond(111) surface grid points.Brown spheres represent C atoms,white spheres represent surface H atoms.
吸附能(Ead)和迁移激活能(Eae)分别定义为
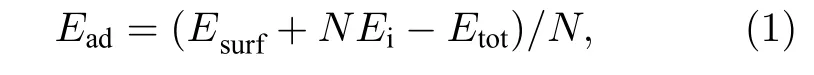
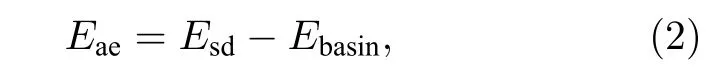
式中,Esurf为表面模型的自由能;N是吸附原子的个数;Ei为吸附原子的单原子能量;Etot为原子吸附后系统的总能量;Esd为迁移路径中鞍点能量,鞍点(SP)是在MEB 上能量最高且其他方向能量最低的位置;Ebasin为迁移路径中的最低能量.
黏附功是衡量界面结合强度的重要物理量,其值越大则表明结合强度越大,其定义是把一个界面分离成两个自由表面在单位面积上所做的可逆功[29].对于金刚石/Al 界面系统来说,可定义为
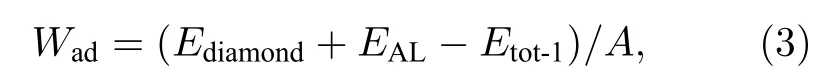
其中,Wad表示黏附功,Ediamond和EAl分别表示金刚石和金属Al 表面的能量,Etot-1表示界面模型的能量,A表示界面面积.
3 结果与讨论
3.1 金刚石晶体的前期优化计算
前期计算包括参数优化和模型验证,其中参数优化包括表面模型层数和真空层的设置、截断能选取、布里渊区k点划分、Smearing 展宽等,具体结果见计算模型部分;为校验金刚石结构参数,采用二次优化方法[30,31]优化其晶格常数(a),并根据Voigt-Reuss-Hill 理论[32−34],验证其弹性常数(C11,C12和C44)和弹性模量(B,E和G),结果如表1所列.
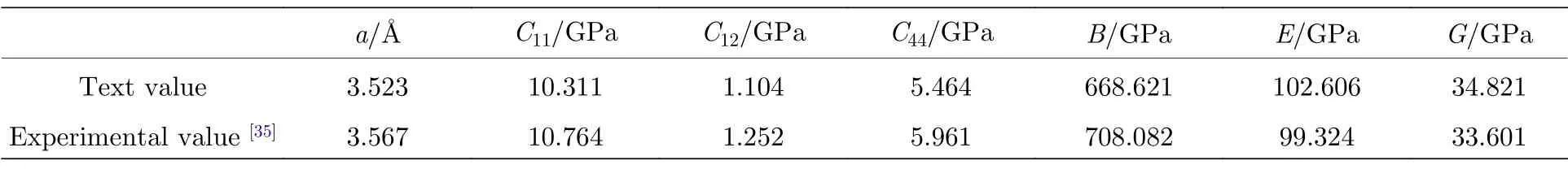
表1 金刚石弹性常数及弹性模量Table 1. Elastic constants and modulus of diamond.
计算结果显示,金刚石晶格常数和弹性模量的计算值与实验值符合得较好,误差基本控制在约5%以内,说明模型和参数选择都是可靠的.
3.2 单个Al 原子的吸附和迁移
对于无过渡区界面,其组成仅有几个原子层,所以单原子的吸附与迁移对界面形成和结构具有非常重要的影响.
根据几何结构分析,金刚石(111)表面的主要高对称位置有Top 位、Br 位、H3 和T4 位置(如图1 所示).为确定Al 原子在H 终止金刚石表面稳定吸附位置,首先在绝热态下确定Al 原子的吸附距离,然后在表面C 原子松弛状态下,对比了上述高对称位置的吸附能,见表2,其中为考虑范德瓦耳斯力作用的吸附能.
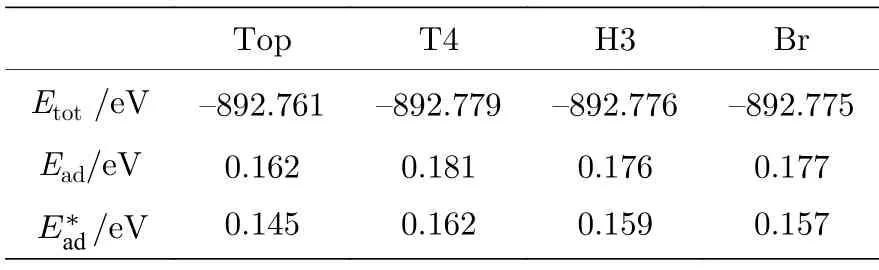
表2 单个Al 原子的吸附能Table 2. Adsorption energies of a single Al atom.
由表2 可知,单Al 原子在H-Ter 终止表面的吸附能都较低,其中吸附能最高的是T4 位置,仅为0.181 eV.并且不同位置间的吸附能相差也不大,最大差值仅为0.019 eV.考虑范德瓦耳斯力作用下,Al 原子的吸附能变得更小,但这种改变对4 种位置作用几乎相同,即对吸附位置的分析是相同的.由于范德瓦耳斯力作用是总体效应,对吸附和界面结构影响趋势不变,故以下的计算不再讨论.
Al 原子在H-Ter 表面的迁移路径如图2 所示.由吸附位置分析,Al 原子的可能迁移路径为由T4-Br 位置,考虑到H-Ter的势能面较为光滑,由T4至Top 和由T4 至H3 迁移也进行了计算.
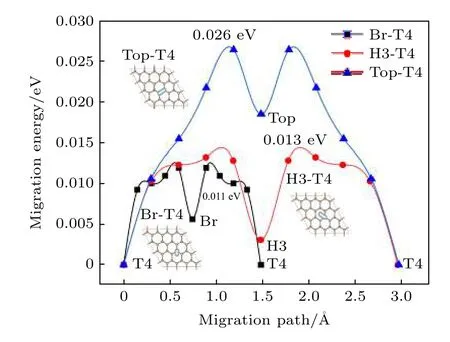
图2 单个Al 原子在H-Ter 表面吸附位置及迁移路径,其中纵坐标为插入点的能量差值Fig.2.Adsorption positions and migration paths of single Al atom on H-Ter surface.The ordinate is the energy difference of the insertion point.
结果显示,在T4 至Br 间,迁移距离最短,迁移激活能也最低(0.011 eV);在T4 至Top 位置间,迁移距离最长,迁移激活能也最高(0.026 eV);而在T4 至H3 位置间的迁移,距离与T4 到Top相近,激活能与T4 到Br 相近(0.013 eV).总体而言,在H 终止金刚石表面Al 原子的迁移较为容易,可认为在微扰动情况下即可实现在Br,H3 和T4位置间的迁移,而在Top 是非稳定位置,这在吸附能和迁移激活能都有所反映.
Al 原子在金刚石表面吸附能低是由于表面C原子与终止H 原子成键,形成饱和键型,使得Al原子主要以物理形式吸附于金刚石表面,这可由Al 和金刚石表面的电荷密度图看出,见图3.
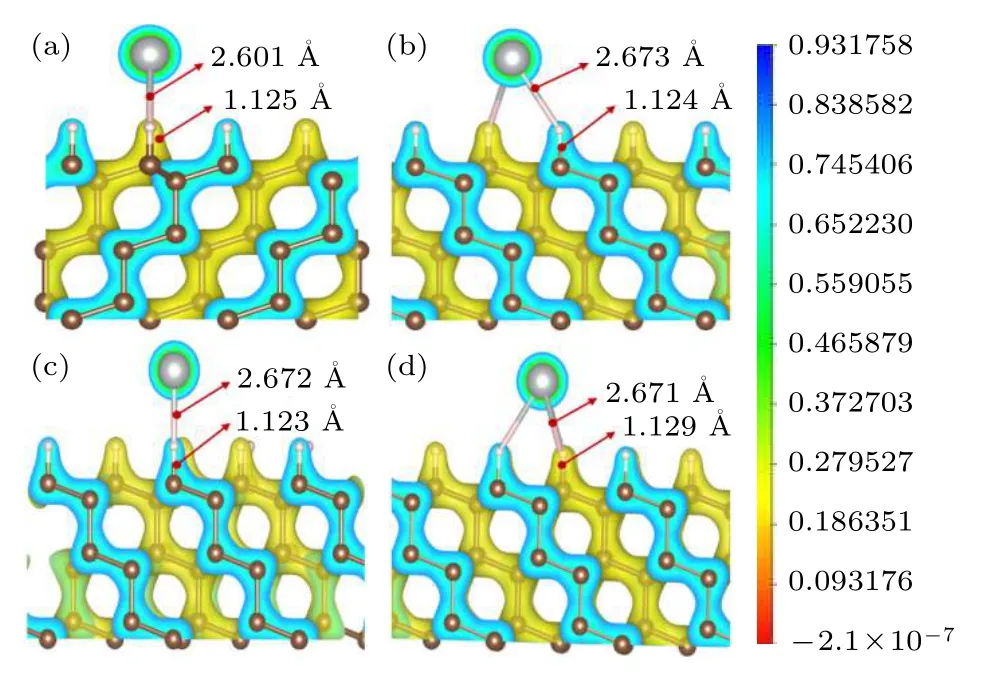
图3 Al 原子在Br(a),H3(b),Top(c),T4(d)吸附位置的电荷密度分布图,图中银白色球为Al 原子,颜色从蓝到红表示电荷密度从高到低Fig.3.Charge density distribution of single Al atom adsorbed on Br(a),H3(b),Top(c),and T4(d) sites,respectively.The big silver ball in the picture is Al atom.The color from blue to red indicates the charge density from high to low.
C—C 间的电荷密度呈现明显四面体分布,与金刚石晶体中sp3杂化键符合得非常好.C—H 间电荷密度发生明显偏移,原子间形成离子键.而Al 原子与金刚石表面几乎没有电荷密度重叠,即Al 原子与晶体表面成键很弱或没有成键,这与表面Al的低吸附能结果分析是一致的.
从原子间距离分析,Al 在Br,H3,T4,Top 吸附位置时,Al—H 原子距离分别为2.601,2.673,2.671 和2.672 Å,而此时表面C 原子与相应的H 原子间的键长为1.125,1.124,1.129 和1.123 Å.对比驰豫后的C—H 键长1.109 Å,可以看出金刚石表面吸附Al 时,表面C—H 键仍受到影响,C—H 键长不同程度地增大.这说明Al—H仍存在较弱作用力.
虽然差分电荷密度也能部分反映出Al—H 间的电荷转移,但由电荷布居分布bader 分析可定量化表面原子的电荷转移量,见表3 所列.表中序号标记为离Al 原子近邻位置,如C1,H1 为Al 最近的邻碳原子和氢原子.
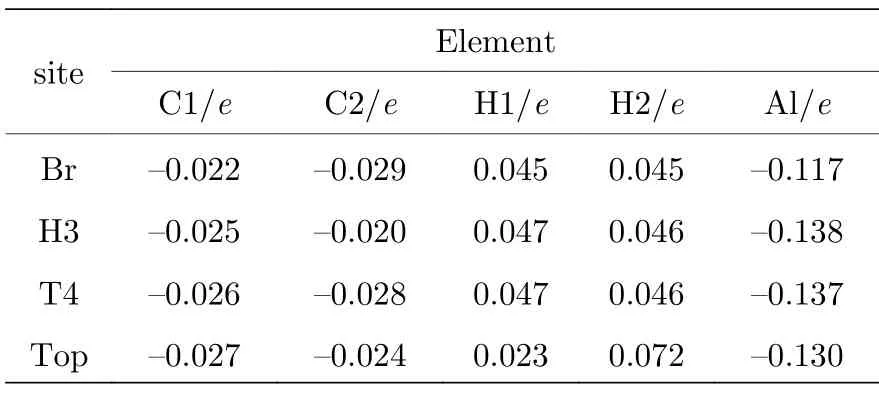
表3 不同吸附结构中Al 原子及其周边原子电荷转移量Table 3. Amount of charge transfer of Al atom and its surrounding atoms in different adsorption structures.
由表3 可知,4 种吸附位置的表面Al 都呈失电子状态,其中H3 和T4 位置失电子最多,这与吸附能的结果趋势是一致的.极性表面使得H 原子比Al 原子更容易获得电子呈现弱的负价态.金刚石表面C 相较于晶体内部C 原子捕获电子能力弱而失去电子.Top 位置较为特殊,虽然Al—H 间电荷转移量较多,但非稳定结构,吸附Al 原子“压迫”与H 相连的C 原子,使系统能量升高.
综上,Al 在H 终止金刚石表面的吸附性能较弱,Al—H 间虽有部分电荷转移,可被认为是静电吸附,所以表面Al 原子为物理性吸附.弱的相互作用使得Al 原子在金刚石表面势能面非常平滑,原子容易发生迁移,且迁移激活非常小.
3.3 金刚石/Al 界面性能
原子Al 在H 终止金刚石表面主要为物理吸附.弱的吸附能和光滑势能面使得吸附Al 原子容易通过简单迁移形成最低能量结构.所以H 终止金刚石与Al 形成的界面结构对表面原子位置并不敏感.那么沉积态Al 薄膜与金刚石基底能否形成稳定的界面,此类界面的性质如何? 表4 列出了由上述3 种吸附位置形成界面的黏附功.
由表4 可知,几种金刚石/Al 界面的黏附功都大于0,表明金属Al 在H 终止金刚石表面理论上是稳定的.相较于其他位置,Top 位置形成的界面黏附功最小,可见即使单个Al 原子可稳定吸附,但铺满整个表面形成界面时,可能由于表面原子起伏等原因,使得界面结构处于亚稳定状态.由于界面的黏附功均在0 附近,界面的稳定性在外部环境改变时极易被破坏,如在高温下,熔化后的Al 液,只需很小表面张力就可超过此界面的黏附功,而形成不润湿界面.
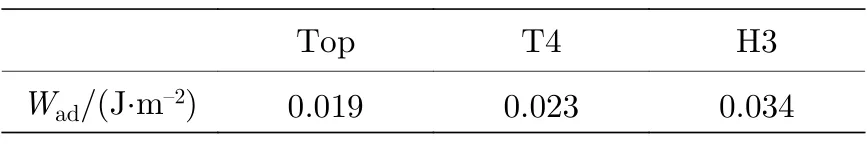
表4 由吸附位置形成界面的黏附功Table 4. Adhesion work of the interface formed by the adsorption position.
对于上述猜测,采用第一性原理分子动力学对界面在温度效应下金属Al 层的演变过程进行了仿真.仿真结果如图4 所示.
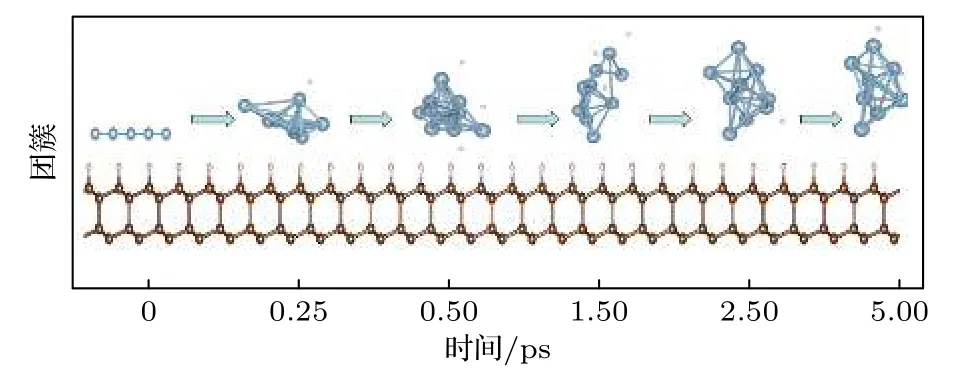
图4 金刚石/Al 界面处Al 液形成过程Fig.4.Formation process of molten aluminum at the diamond/Al interface.
简单模拟可知,在高温下Al 原子层逐渐有原子脱离界面向Al 层上侧运动,在这一运动过程中可能“裹挟”部分少量H 原子,这一过程持续至仅有少量Al 原子与金刚石界面接触(此模型下仅有1 个Al 原子),最终形成的Al 团簇为两端(界面端和真空端)原子较少,中间原子较多的形状.可初步认为这是金属Al 熔化后,在表面张力作用下Al 原子收缩形成类液滴状.这与Smirnov 等[3]实验现象是相符的.在退火过程中,Al 团簇间原子间距开始收缩,在约300 ℃时接近稳定,Al 原子间的距离不再有太大变化,Al 团簇结构也不再变化.这说明Al 液固化后,原子位置维持状态不再变化.整个过程中,金刚石原子几乎不动.分子动力学仿真结果基本上与实验现象相符,验证了上述关于金刚石/Al 界面不稳定的猜测.
不稳定的金刚石/Al 界面,仍存在原子间相互作用力.图5 是界面的态密度图投影显示(PDOS),PDOS 中并未发现界面Al 原子和H 原子间产生轨道杂化的共振峰,再次说明Al—H 间虽有少量电荷转移,但并形成化学键.界面处C 原子2s2p 和H 原子1s 轨道产生了大量的界面带隙态.界面附近第1 层C 原子2p 轨道未有因金属诱导而产生明显的带隙态,而氢化表面的H 原子1s 轨道对金属诱导带隙态提供少量贡献.除此之外,第2 层C 受到的界面影响大大减弱.第3 层C 原子的态密度逐渐恢复到半导体特性.这说明金属诱导带隙态在半导体氢化金刚石一侧产生微弱的局域化特征.
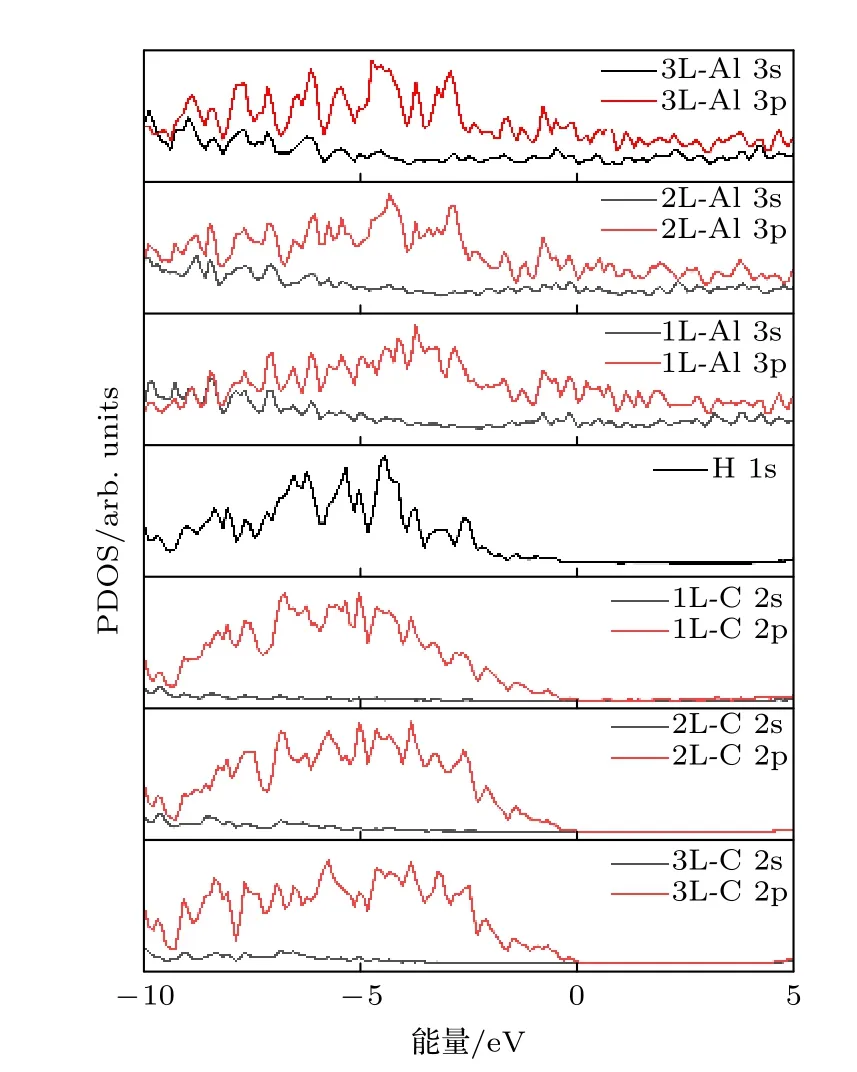
图5 金 刚石/Al 界 面的PDOS 图,图中的L 表示距离界面远近的C 原子层数Fig.5.PDOS diagram of metal aluminum-diamond interface.The L indicates the lay of C atom far from the interface.
这也就容易理解,Smirnov 等[3]的实验原理.沉积态Al 薄膜物理吸附于金刚石表面,吸附能力弱,粒子表面活动能力强.所以Al 薄膜对H 终止金刚石表面原子结构并不敏感.薄膜沉积过程能量损失速度高达1013K/s[29],高能态原子可形成结构并不稳定的金刚石/Al 界面,这种界面存在少量的电荷转移,有较弱的界面作用力.金属掩膜纳米球的形成就是由不稳定的金刚石/Al 界面在得到能量后(热处理条件提供),在重力和表面张力的作用下,沿金刚石表面形貌形成的规则不一的金属球团.所以,这种金属纳米掩膜,对金属与基底材料作用关系要求不高,可能主要取决基底材料的表面形貌.金刚石与大多数金属的润湿性都不好,加之沉积金刚石的H 终止表面的隔离作用,可能许多金属都可具备上述金属纳米掩膜的形成条件.所以精确控制金刚石薄膜表面形貌有望发展出一种简单有效的金属纳米掩膜控制方法.
4 结 论
本文采用第一性原理计算研究了单个Al 原子在H 终止金刚石表面的吸附及其迁移行为,分析了金刚石/Al 界面的结构与性能:
1) Al 原子在H 终止金刚石表面吸附时,对表面原子位置不敏感,几个高对称的吸附能几乎相同(约0.2 eV).吸附Al 原子的表面势能面也非常平滑,迁移激活能非常小(约0.01 eV).
2) 通过电荷密度、电荷布居和原子间距分析,Al 与H 原子间没有形成化学键,仅有少量电荷转移,所以Al 原子在H 终止金刚石表面是物理吸附.
3) 由沉积Al 原子与H 终止金刚石形成的金刚石/Al 界面不具有能量稳定性,在外界高温环境下可形成与金刚石表面形貌相关的金属纳米球.
本文结果为理解金属纳米掩膜的形成机理提供重要的理论参考.
猜你喜欢
导航定位学报(2022年5期)2022-10-13
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
人工晶体学报(2021年3期)2021-04-17
粉末冶金技术(2021年1期)2021-03-29
石材(2020年10期)2021-01-08
石材(2020年7期)2020-08-24
石材(2020年2期)2020-03-16
电子技术与软件工程(2018年5期)2018-04-09
