
我国药品抽检质量风险提示函制度的探讨
2021-11-24 18:06刘文王翀朱炯胡增峣
中国药房 2021年21期
刘文 王翀 朱炯 胡增峣


中图分类号 R951 文献标志码 A 文章编号 1001-0408(2021)21-2575-06
DOI 10.6039/j.issn.1001-0408.2021.21.03
摘 要 目的:为进一步完善我国药品抽检质量风险提示函(以下简称“提示函”)制度、加强药品质量管理水平提供参考。方法:概述我国药品抽检的质量风险管理情况,对其中提示函的法律性质、主要内容和工作程序进行分析,以最新的2020年的提示函数据为例分析提示函在排查潜在药品质量风险中的作用,并针对目前存在的问题提出完善建议。结果与结论:在基于法定标准检验的同时,我国药品监督管理部门对可能因生产企业的药品质量控制盲区或偏差而存在质量安全风险的药品开展了探索性研究,并按照发现问题的严重程度分为严重风险和一般性风险,实施分级管理。提示函就是基于劝服优先的原则对一般性药品质量风险制定的行政措施,不具有制裁性。其主要内容涵盖风险排查整改所需全部信息(药品基本信息、提示的风险信息、联系人信息、发现风险的方法、排查整改要求、属地省级药监部门的职责),涉及承检机构、中国食品药品检定研究院、国家药监局、相关生产企业属地省级药监局与相关生产企业共五方责任主体,通过闭环管理的模式实现监管部门与生产企业之间的良性互动。2020年,国家药监局共向286家生产企业发出312份提示函,提示准确率达87.91%,具有较好的科学性和针对性。生产企业针对提示函的内容进行了包括开展工艺验证、修订内控标准、加强生产过程控制等一系列整改措施,但也存在提示内容的合理性遭到生产企业质疑以及生产企业排查力度不足等问题。鉴于此,笔者建议生产企业要正确认识提示函的性质和价值,承检机构应进一步提高发现问题的科学性和针对性,而药监部门则要重点关注排查发现的问题,以共同促进药品质量的提升。
关键词 药品抽检;風险管理;提示函;劝服优先
ABSTRACT OBJECTIVE: To provide a reference for further improving the quality risk reminder mechanism of the drug sampling and testing (called “the Reminder” as for short) in China, and strengthening the drug quality management. METHODS: The quality risk management situation of the drug sampling and testing were summarized, and the legal nature, main content and working procedures of the Reminder were analyzed. The latest data of the Reminder in 2020 were taken as an example to analyze the role of the Reminder in the investigation of potential drug quality risks, and made the suggestions for existing problems. RESULTS & CONCLUSIONS: Based on legal standards, Chinas drug regulatory departments had carried out exploratory research on drugs that may have quality and safety risks due to drug quality control blind spots or deviations of manufacturing enterprises, and divided them into serious risks and general risks according to the severity of the problems found, and implemented hierarchical management. The Reminder was an administrative measure for general drug quality risks based on the principle of persuasion first, and did not have sanctions. Its main content covered all the information required for risk investigation and rectification (basic drug information, suggested risk information, contact information, risk discovery methods, troubleshooting and rectification requirements, and responsibilities of local provincial drug regulatory departments). It involved five responsible parties, ie. the inspection institution, China Institute for Food and Drug Control, National Medical Products Administration, the provincial food and drug administration of the relevant manufacturing enterprises and the relevant manufacturing enterprises. Through the mode of closed-loop management, the benign interaction between regulatory authorities and manufacturing enterprises could been realized. In 2020, National Medical Products Administration issued 312 reminders to 286 manufacturers, with an accuracy of 87.91%, which was scientific and targeted. The manufacturer had carried out a series of rectification measures for the contents of the Reminder, including carrying out process verification, revising internal control standards and strengthening production process control. However, there were also some problems, such as the rationality of the prompt contents being questioned by the manufacturer and the insufficient investigation of the manufacturer. It is suggested that the manufacturers correctly understand the nature and value of the Reminder. The inspection agency should further improve the scientific pertinence of the problems found, while the drug regulatory department should focus on the troubleshooting of the problems found, so as to jointly promote the improvement of drug quality.
KEYWORDS Drug sampling and testing; Risk management; Reminder; Persuasion first
行政执法中的一个重要原则是劝服优先(persuasion first),是指在行政执法中首先考虑以劝导模式实现规制遵从,即以强制力较弱的监管措施促使被监管者主动建立内部合规体系,这样可以合理配置监管资源、降低执法成本,有效实现监管目标[1]。《行政处罚法》规定,“实施行政处罚和纠正违法行为应坚持处罚与教育相结合,教育公民、法人或者其他组织自觉守法”;《药品管理法》也规定了“对于尚不影响药物安全性和有效性的轻微违规行为,应责令限期改正”[2]。
借鉴这一原则,我国于2014年建立了药品质量风险提示函(以下简称“提示函”)制度,即药品监督管理部门对国家药品抽检探索性研究中发现的尚不影响药物安全性和有效性的一般性风险,向相关生产企业发出劝导性告知,要求其开展排查并采取必要的整改措施[3-4];生产企业据此采取优化工艺、加强生产过程管理、完善标准等措施,不断提升药品质量管理水平,主动落实药品质量安全的主体责任。提示函从源头上提高药品质量方面发挥的作用愈加显著,已成为我国药品监管的重要策略。为进一步加强和完善我国提示函制度,笔者结合多年工作经验,对提示函制度体系进行研究,以期为有关部门提供参考、为加强药品质量管理提供借鉴。
1 国家药品抽检风险管理概述
人用药物注册技术要求国际协调会(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)于2005 年发布《质量风险管理》(Quality Risk Management)的第Q9号技术文件,要求对药品的整个生命周期进行质量风险管理(risk management)[5],即采取药物警戒行动和干预等措施,识别、预防和减少药品相关风险。这是对整个产品周期全面和持续降低风险的过程,旨在实现效益风险最优化[6]。近年来,我国国家药品抽检紧密结合监管需要,积极改革体制机制,借鉴ICH的理念加强药品风险管理,提出了一系列创新性举措,加大、加深、加强对假冒伪劣药品和潜在质量安全风险的挖掘和处置,主要是在基于法定质量标准检验的同时,对可能因生产企业的药品质量控制盲区或偏差而存在质量安全风险的药品开展探索性研究[7],重点排查工艺处方、原料、生产过程、辅料、包装材料(以下简称“包材”)等可能影响药品内在质量的相关因素,为进一步提升药品质量水平、加强药品监管提供技术支持。
为合理配置药品监管资源、提高风险管理效率,国家药监局对于国家药品抽检探索性研究中发现的问题,按照严重风险和一般性风险实施分级管理。严重风险主要是国家药品抽检承检机构经分析,认为探索性研究发现的对公众用药安全有效的不利影响较大或涉嫌主观故意、违法违规而必须立即采取控制措施的问题[8];其管理措施主要是以飞行检查或现场核查的形式对相关生产企业进行检查,查实违法违规行为时依法依规立案查处。一般性风险即国家药品抽检承检机构经分析,认为探索性研究发现的安全风险较低或非主观故意、未涉嫌违法违规的问题[8];其管理措施主要是药监部门以提示函的形式告知生产企业并提供检验方法,帮助生产企业主动建立内部合规体系、提高藥品质量。国家药品抽检具体实践中发现一般性风险的发生率高于严重风险。加强一般性风险的管理,避免其进一步恶化或升级成为严重风险,与严重风险管理一样,也对提升药品整体质量水平、保护公众用药安全有效具有重要意义。
2 提示函的法律性质分析
如上所述,我国的提示函适用范围是国家药品抽检探索性研究中发现的一般性问题,并且相关生产企业可以纠正偏差或消除风险,从而由国家药监局向相关生产企业发出提示函并提供检验方法供其无偿使用。这与有证据证明假冒伪劣药品的核查处置以及上述严重风险的有因检查等风险管理措施显著不同。提示函是根据劝服优先的原则向相关生产企业发出的通知,目的是要求企业对提示的问题开展排查整改,而不涉及《药品管理法》规定的药监部门对有证据证明可能存在安全隐患的药品采取告诫、约谈、限期整改以及暂停生产、销售、使用、进口等制裁性措施。因此,提示函不是处罚手段,不具有制裁性。另一方面,提示函向相关生产企业提供检验方法充分体现了我国药监部门帮扶生产企业、寓服务于监管的作风。例如2020年国家药品抽检中,承检机构在探索性研究中发现复方托吡卡胺滴眼液中存在环氧乙烷与托吡卡胺反应生成的未知杂质,并建立了该品种中托吡卡胺环氧乙烷化合物检查方法,通过提示函的形式提示有关生产企业登录中国食品药品检定研究院(以下简称“中检院”)官网,在相应模块中下载使用检验方法以加强对该杂质的控制。
提示函与美国食品药品管理局(FDA)的无标题信(untitled letters)类似。美国FDA的无标题信也是针对药品监管过程中发现的一般性违规行为,根据劝服优先原则向相对人发出的通知,以此对相对人进行提醒,以这种非处罚性的、强制力较弱的监管手段促使生产企业采取相应的措施,避免相同或类似行为再次发生[9]。其与我国提示函主要的不同在于美国FDA在其官方网站公开无标题信但不公开相对人的姓名或生产企业名称,以“广而告之”的形式要求相对人及时纠正违规行为;而我国的提示函发出后,国家药监局会要求属地省级药监部门结合日常监督检查,有针对性地加强对相关生产企业的风险管理,以此达到约束相关生产企业执行监管部门要求的效果。
综上所述,我国的提示函不是对相关生产企业的处罚手段,而是针对其药品生产过程中出现的偏差,以技术支持的方式实行的干预措施。这贯彻落实了《药品管理法》关于药品管理应满足风险管理、全程管控、社会共治的要求,帮助药品上市许可持有人开展药品上市后研究,对药品的安全性、有效性和质量可控性进行进一步确证,加强对已上市药品的持续管理。
3 提示函的主要内容
为促进相关生产企业充分理解提示函的要求,便于其采取针对性的排查整改措施,提示函应涵盖风险排查整改所需全部信息,主要包括以下内容。
3.1 药品基本信息
提示函中的药品基本信息包括国家药品抽检探索性研究中发现存在一般质量风险的药品的通用名、生产企业及所在省份、药品批号等。通过这些信息可以帮助相关省级药监局锁定被提示的生产企业,并指导该生产企业锁定需开展风险排查的目标药品及其生产过程。
3.2 提示的风险信息
提示函的主体内容即为提示的风险信息,包括对探索性研究中发现的一般质量风险内容、发现的途径和现象,并基于探索性研究数据对可能造成这一风险的原因进行初步分析,以简要但足够详细的事实描述为相关生产企业的风险排查提供参考。
3.3 联系人信息
为便于生产企业在需要时针对具体问题与承检机构进行技术沟通,提高风险排查整改的效率,提示函中会提供发现相关风险的承检机构联系人及联系方式,同时也要求承检机构认真解答相关生产企业的咨询。
3.4 发现风险的方法
承检机构发现风险所用的方法是提示函的另一项重点内容,包括承检机构探索性研究的新建方法、参照其他药品的方法或非实验性方法。在提示函中告知生产企业这些方法的正文已通过中检院官网公开,可自由下载[10],便于相关企业在需要时使用。
3.5 排查整改要求
提示函的目的即为相关生产企业对风险进行排查整改,因此提示函中会要求相关生产企业在规定时间内围绕所提示风险认真排查可能存在的隐患,对确实存在问题的隐患及时采取整改措施并向属地省级药监部门报告。
3.6 属地省级药监部门的职责
国家药监局要求属地省级药监局将提示函送达相关生产企业,督促指导企业针对提示的问题开展排查整改,结合日常监督检查工作有针对性地加强对企业的风险管理,并汇总整理各相关生产企业的排查整改情况后报告给国家药监局。
4 提示函的工作程序
按国家药监局组织制定的工作程序,提示函涉及承检机构、中检院、国家药监局、相关生产企业属地省级药监局、相关生产企业等五方责任主体,工作过程包括数据征集、专家研判、提示函发送、排查整改和结果反馈等5个阶段,具体如下:
承检机构在国家药品抽检探索性研究中发现一般性风险后整理数据和方法并按要求报送中检院,中检院组织相关领域的检验专家对所报风险进行函审。如函审提出不同意见,则中检院及时反馈给承检机构进行修改;然后中检院再次组织专家以现场会议的形式作出进一步研判,提出具体的处置建议。中检院根据专家会议决议整理相关数据后报送国家药监局并公开其所用方法。国家药监局部署相关生产企业属地省级药监局组织相关生产企业排查整改。属地省级药监局将提示函送达相关生产企业,告知排查整改的要求及书面报告结果的时限,并提供力所能及的帮助;而在收到相关生产企业的排查整改情况汇报后及时报告国家药监局,并在下一步的工作中重点关注相关生产企业产品的质量状况,必要时加强监管。国家药监局组织中检院将排查整改的情况反馈给承检机构,供承检机构在下一步的工作中参考。以此形成闭环管理模式,实现监管部门与生产企业之间的良性互动,如图1所示。
5 提示函制度实施情况分析
本文对2014-2020年提示函的整体情况进行分析,并对最新的2020年提示函总体情况、排查情况和整改情况、排查后仍存在的問题等进行分析,结果如下。
5.1 2014-2020年提示函整体情况
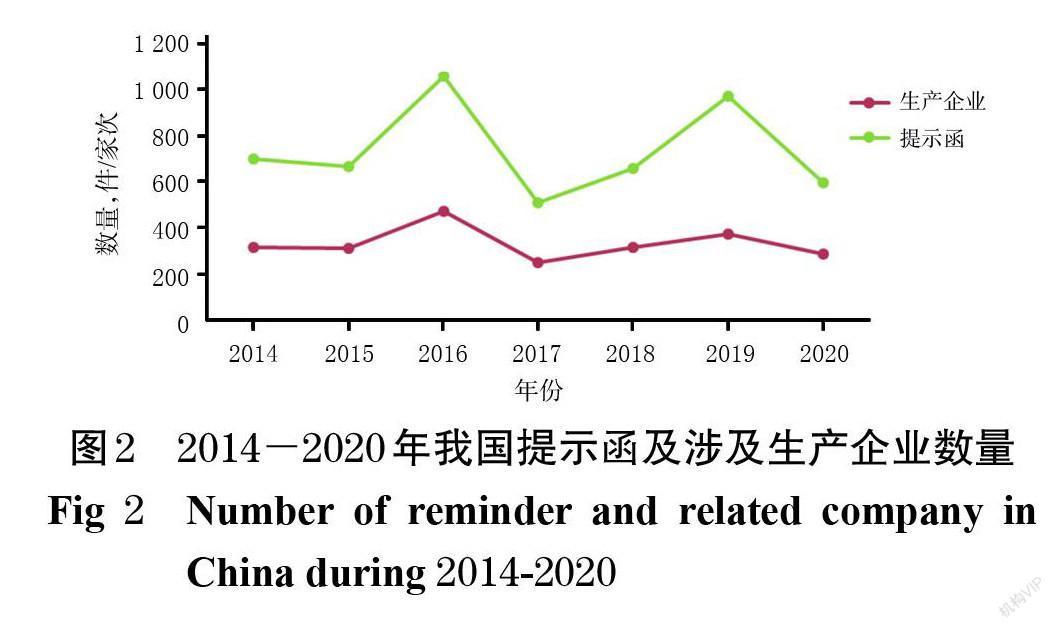
2014-2020年,国家药监局已向2 323家次的生产企业发出2 837份提示函,涉及化学药、中成药、中药饮片、生物制品、包材、辅料等各个类别的产品。由于每年国家药品抽检的具体药品不同,其质量状况各不相同,因此每年的提示函数量及被提示生产企业数量也随之有所变化,如图2所示。
5.2 2020年提示函整体情况
2020年,国家药监局向28个省(区、市)及新疆生产建设兵团的286家生产企业发出312份提示函,主要涉及化学药、中成药、中药饮片、血液制品和包材等51个品种的质量风险,整体情况如表1所示。提示函提示的质量风险主要包括在生产工艺控制、原料、辅料、包材、说明书、标签标识等方面存在的缺陷或不足,承检机构探索性研究发现这些问题的方法也在中检院官网进行了同步公开。
5.3 2020年排查情况
各属地省级药监部门按提示函要求组织生产企业就提示函中的风险信息积极开展排查、分析和验证并将排查整改情况反馈给国家药监局。2020年,312份提示函中有6份中药饮片的提示函由于涉及的6家生产企业已注销,不会继续生产药品或继续产生风险,故无排查必要。因此,在产的280家生产企业对306份提示函中的问题开展了排查,排查情况如表2所示。其中,以生产企业根据提示函对药品生产全过程各环节开展排查并找到被提示的问题视为准确提示。从排查结果来看,各类药品排查发现问题的提示准确率均超过85%,整体准确率达87.91%,说明2020年提示函具有较为满意的科学性和针对性。
5.4 2020年整改情况
通过省级药监局反馈的生产企业整改情况可以发现,各生产企业均开展了问题排查分析,查找原因并采取了具有针对性的多项整改措施,主要措施如下。
5.4.1 开展工艺验证 提示函所提示的风险经排查发现生产工艺控制不佳者,其整改措施包括开展工艺验证,必要时按要求申报变更。例如针对提示函提示的阿魏酸哌嗪片乙醇残留量超标的问题,某生产企业的整改措施包括开展阿魏酸哌嗪片控制乙醇残留颗粒预干燥时间和进风温度的工艺验证。
5.4.2 修订内控标准 提示函所提示的风险经排查发现内控标准存在不足者,其整改措施包括对内控标准进行提高或完善。例如针对提示函提示的红金消结胶囊缺少石细胞特征的问题,某生产企业的整改措施包括修订成品内控质量标准,增订石细胞鉴别项及鬼臼毒素含量测定控制项。
5.4.3 加强生产过程控制 提示函所提示的风险经排查发现生产过程控制不当者,需对其生产过程加强控制。例如针对提示函提示的安神补脑胶囊中的制何首乌、淫羊藿、甘草等原药材指标性成分的转移率较低的问题,某生产企业的整改措施包括加强中药提取生产过程的管理,严格执行中药提取过程药材的拣、洗、润、切、干燥等前处理工序,保证中药炮制后的产品质量,加强对中药提取、浓缩、醇沉、真空干燥等生产工序的工艺参数的监控。
5.4.4 加强“原、辅、包”质量控制 提示函所提示的风险经排查发现与原料药、辅料或包材有关者,其整改措施包括加强对原料、辅料或包材的质量控制。例如针对提示函提示的注射用头孢尼西钠原料可能带入异亚丙基丙酮杂质的问题,某生产企业的整改措施包括暂停使用某原料药企业生产的头孢尼西钠原料用于制剂生产,并建立异亚丙基丙酮杂质检测的方法与限度标准,同时对其他供应商原料生产工艺进行排查。
5.4.5 申请修订药品说明书 提示函所提示的风险经排查发现药品说明书中存在缺陷或不足者,其整改措施包括申请修订药品说明书。例如针对提示函提示的复方感冒灵颗粒说明书中化学药成分的安全性信息不完善的问题,某生产企业的整改措施包括对复方感冒灵颗粒近5年来的安全性信息进行评价,在药品说明书中增加不良反应、用药禁忌和注意事项等内容,现已完成补充申请备案。
5.4.6 加强人员培训 提示函所提示的风险经排查发现生产人员的知识技能不足者,需加强对其相关人员的培训。例如针对提示函提示的防风饮片薄层色谱中特征斑点较弱的问题,某生产企业的整改措施包括安排检验经验丰富的人员对采购、验收人员进行培训,对防风栽培品与野生品如何鉴别等知识进行系统学习。
除上述整改措施外,大部分生产企业为控制药品风险、减少用药安全隐患,召回了已上市的药品;也有生产企业在整改期间暂停相关品种的生产,待整改到位后恢复生产。以上情况充分说明,提示函可以有效规范企业的生产行为,促进企业生产管理水平的提升,进而防控潜在的药品质量安全风险。
5.5 2020年排查存在的问题
通过排查情况可以发现,仍有27家生产企业的37份提示函经相关生产企业排查未能找到生产过程中存在的相应问题,分别占生产企业总数的9.64%(27/280)、占提示函总数的12.09%(37/306),涉及除血液制品外的所有类别的药品,提示的问题则涵盖了工艺处方、原料、辅料、药品包材、说明书、标签标识等各个方面。主要问题包括:(1)生产企业质疑提示风险的合理性——部分生产企业从中检院官网下载相关品种的方法后开展研究,认为承检机构的方法限度设置过高、无法重现承检机构的检验结果、企业实验室不具备承检机构的实验条件等,也未按提示函中所示的联系方式与承检机构进行沟通,即认为自身在药品生产过程中不存在问题;(2)生产企业排查力度不足——部分生产企业收到提示函后仅对批生产记录、批检验记录、物料购进记录、投料监控记录等进行了检查,认为符合现行版《药品生产质量管理规范》(Good Manufacture Practices,GMP),却忽略了这些记录无法体现物料、人员、设施、设备、工艺、处方等所有可能影响药品质量的细节问题,也未按中检院官网公开的方法开展样品检验研究。 这些问题都有待进一步解决。
6 讨论
通过以上分析可以发现,当前我国化学药、中成药、中药饮片、生物制品和包材的生产工艺、内控标准、原料、辅料、包材、生产过程、说明书等方面仍然存在诸多一般性风险。2014年至今,国家药品抽检探索性研究挖掘出了其中大量的问题,通过提示函使生产企业进行排查并采取有效的、有针对性的整改措施,以较高的科学性和针对性指导生产企业发现并解决问题,进一步规范药品生产全过程,促进制药行业健康发展,促进潜在用药安全隐患的防控和药品质量水平的持续提升。提示函这一药品监管的创新性策略,也是对药品监管科学理论的践行和发展。
此外,药监部门也应该认识到具体工作中仍存在诸多问题,不利于充分发挥提示函的作用。因此,为进一步加强和完善提示函制度、不断提高药品质量水平,笔者提出以下建议。
6.1 生产企业要正确认识提示函的性质和价值
部分生产企业向属地省级药监局反馈的排查整改情况中过度强调GMP合规,明显带有撇清责任的意思,由此可以发现企业对提示函的认识有待加强。生产企业应正确认识提示函是基于劝服优先原则的提醒,不具有制裁性,是承检机构利用国家药品抽检的资源帮助生产企业发现并控制药品质量风险的举措。另一方面,美国FDA认为药品生产企业不能仅仅满足于GMP合规,GMP合规只是一个最低限度的基本要求,并不能说明生产工艺和管理体系不存在任何瑕疵;药品生产企业若仅满足于GMP合规,会失去提升药品质量管理能力的机会,因而FDA鼓励药品生产企业主动采取措施不断加强和完善药品生产工艺和管理体系,积极与药品监管机构进行沟通[11-12]。同理,我国的药品生产企业也不能仅仅满足于GMP合规,而应本着切实对药品质量担负主体责任的态度,正确理解提示函的性质及其对提高药品质量的作用,消除对提示函的抵触情绪,充分借助提示函中的问题提示及检验方法深入排查生产过程中可能影响药品质量的风险,遇到问题时积极与承检机构开展技术交流和沟通,不断提高自身的药品质量控制水平。
6.2 承检机构进一步提高发现问题的科学性和针对性
药品检验是通过科学的方法评价药品质量特征、排查潜在风险,其准确性、规范性和公正性直接影响到检验机构的公信力[13-14]。对于部分生产企业排查未发现问题的情况,承检机构也应认真反思报告的药品质量风险的科学性,认真研究中检院反馈的生产企业排查整改情况,在下一步的工作中对于成功的经验要加以推广和复制,对存在的缺陷和不足则要修正和改进。在以后的国家药品抽检探索性研究中,笔者建议承检机构充分借助现有技术条件对抽检品种的生产全过程开展调研,找到可能影响药品质量的關键因素;在此基础上设计详实细致的探索性研究方案,建立科学合理的检验方法并进行严格的方法学验证,加强探索性研究工作的质量管理,实现方法科学、数据准确;并将药品质量风险的研究数据置于药品工业化生产的背景中进行分析,将研究数据与生产企业的生产经营活动相关联,识别其中存在的缺陷和不足,务必排除误判的可能性,不断提高提示函的科学性和针对性。
6.3 药监部门重点关注排查发现的问题
针对生产企业根据提示函排查发现的问题,例如化学药和包材中较为突出的工艺控制问题、中成药的原料药材质量问题、中药饮片的炮制规范性问题等,建议属地药监部门继续指导和帮扶相关生产企业整改到位,确保风险得到有效控制。另一方面,药监部门要加强对上述问题的日常监管,将其列入重点检查的内容。若药监部门在后续检查中发现相关生产企业未建立内部合规体系致使同样的问题再次出现,则应采取强制力较强且具有制裁性的监管措施。例如药监部门可借鉴美国FDA基于风险的检查场地选择模型(site selection model),将相关生产企业未按无标题信要求对药品质量风险排查整改到位的情况视为优先检查的风险因素[15],在后续日常监管中缩短对其检查的时间间隔并开展重点检查;发现问题时,继续借鉴美国FDA无标题信的形式对其违规行为进行警告并对外公布,以在客观上对其声誉产生不利影响,达到惩戒的目的;后期根据相关生产企业的整改情况,决定撤销警告或采取具有强制力、制裁性、威慑力的监管措施[16]。
参考文献
[ 1 ] 卢超.社会性规制中约谈工具的双重角色[J].法制与社会发展,2019,25(1):146-163.
[ 2 ] 宋华琳,刘炫.美国FDA警告信的制度架构及启示[J].中国食品药品监管,2019(12):28-35.
[ 3 ] 国家食品药品监督管理总局.国家药品监管总局发出警示函 督促药品生产企业排查风险提升药品质量[EB/OL]. (2014-12-01)[2021-04-20]. https://www.nmpa.gov.cn/directory/web/nmpa/yaopin/ypjgdt/201412011034017- 64.html.
[ 4 ] 朱炯,王胜鹏,刘文,等.国家药品抽检药品质量提示数据分析与探讨[J].中国药事,2020,34(8):909-915.
[ 5 ] ICH. ICH Q9:quality risk management[EB/OL].(2009-10- 03)[2021-04-21].http://www.ich.org/cache/compo/276- 254-1.html.
[ 6 ] 李幼平,文进,王莉.药品风险管理:概念、原则、研究方法与实践[J].中国循证医学杂志,2007,7(12):843-848.
[ 7 ] 刘文,朱炯,胡骏,等.《药品质量抽查检验管理办法》与《药品质量抽查检验管理规定》中检验和复验内容的对比分析[J].中国药房,2020,31(14):1665-1670.
[ 8 ] 刘文,朱炯,王翀.国家药品抽检风险管理主要举措分析与建议[J].中国药学杂志,2020,55(16):1394-1398.
[ 9 ] FDA. Issuance of untitled letters[EB/OL]. [2021-04-20]. https://www.fda.gov/inspections-compliance-enforcement- and-criminal-investigations/compliance-actions-and-acti- vities/issuance-untitled-letters.
[10] 朱炯,刘文,王胜鹏,等.我国药品抽检结果信息公开现状分析与建议[J].中国药学杂志,2020,55(18):1553-1558.
[11] FDA. Quality metrics for drug manufacturing[EB/OL].(2019-01-21)[2020-04-22]. https://www.fda.gov/drugs/pharmaceutical-quality-resources/quality-metricsdrug- manufacturing.
[12] 张景辰.加强事中监管,促进药品产业的供给侧改善:FDA质量量度计划的启示[J].中國食品药品监管,2016(4):56-58.
[13] 王亚龙,王燕玲,张永.药品检验过程中质量控制的必要性分析及有效措施探索[J].名医,2019(2):24-26.
[14] 章莹,孙国君,胡英.食品药品检验研究院公信力影响因素与评价指标研究[J].中国药房,2017,28(13):1746- 1749.
[15] FDA. Regulatory procedures manual,chapter 4:advisory actions[EB/OL]. [2021-04-22]. https://www.fda.gov/media/71878/download.
[16] FDA. Chapter 3:center for drug evaluation and research [EB/OL]. [2021-04-22]. https://www.fda.gov/media/729- 59/download.
(收稿日期:2021-04-28 修回日期:2021-08-09)
(编辑:刘明伟)
