
液相色谱-串联质谱测定鸡蛋中氯霉素残留及北京地区膳食暴露风险评估
2021-11-25 12:20周一卉李香月商方方王涛谷旭
甘肃农业大学学报 2021年5期
周一卉,李香月,商方方,王涛,谷旭
(1.北京市石景山区疾病预防控制中心,北京 石景山 100043;2.中国农业科学院饲料研究所,北京 100081)
氯霉素(chloramphenicol,CAP,图1),是一种酰胺醇类广谱抗生素,抗菌效果好且价格低廉,我国从20世纪90年代将其作为饲料添加剂加入动物饲料中防治动物疾病,用于提高畜牧养殖业的产量[1-3].随着氯霉素临床的广泛应用,再生障碍性贫血、胃肠道不适、免疫力下降等由氯霉素引起的一系列不良反应的报道日渐增多[4-5].2002年,西南农业大学相关研究表明,蛋鸡在多剂量内服氯霉素后,在鸡蛋中的药动力学特征为易吸收,因此在鸡蛋中易形成残留[6],一旦通过食物链的传递,最终进入到人体内形成富集,将不同程度地对食用者的身体健康造成损害.
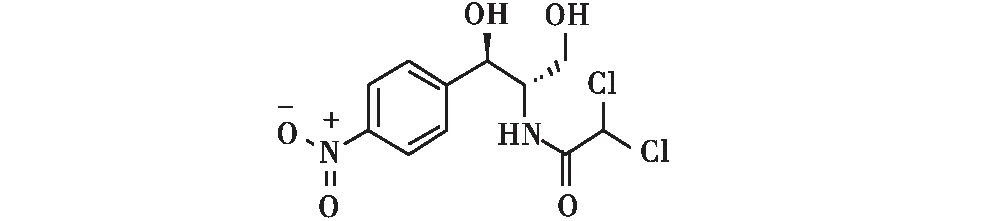
图1 氯霉素结构
日本、欧盟、美国等发达国家都已经将氯霉素列为禁用药物.随着检测技术的不断发展,氯霉素的检出限也逐步降低[7].欧盟在96/23/EC指令中禁止在动物上使用氯霉素,动物源性产品中氯霉素的最高残留限量(MRL)为10 μg/kg[8],检测方法设定最低要求性能限定(MRPL)为0.3 μg/kg[9].2019年,农业农村部公布第250号公告《动物性食品中兽药最高残留限量》中规定,养殖动物禁止氯霉素使用,不得在动物性食品中检出[10].目前,针对动物源性食品中氯霉素检测方法[11]有酶联免疫吸附法、分子印迹法、分子信标法、以及被广泛应用的气相色谱串联质谱法和高效液相色谱串联质谱法等.叶江雷等[12]应用免疫胶体金法检测水产品中氯霉素残留,就如何降低“假阴性”率和提高可靠性,以及如何改进前处理过程提出了建议;张慧洁等[13]建立以氯霉素为模板分子,氧化石墨烯为载体,在碳量子点中掺杂氮、硫元素,采用沉淀法制备氯霉素分子印迹聚合物,并作为荧光探针,对氯霉素残留进行检测,检出限为6×10-4μmol/L.
鸡蛋蛋液中含有大量蛋白质、脂类,相较其他组织鸡蛋基质更为复杂,一方面质谱检测基质效应较强,另一方面目标物与蛋白质相结合难以解离,降低提取效率.氯霉素虽然已经禁止用于动物性食品,但是现行的国标检测方法均是针对动物肌肉组织,还未见关于禽蛋中氯霉素的检测方法.基于HPLC-MS/MS方法灵敏度高,定性准确的特点[14],本研究建立并优化禽蛋中氯霉素残留的固相萃取-超高效液相色谱-串联质谱(SPE-UPLC-MS/MS)分析检测方法,以期提高鸡蛋中氯霉素残留风险监测方法准确度和精密度,为鸡蛋中氯霉素膳食暴露进行风险评估提供依据.
1 材料与方法
1.1 仪器
ACQUITYTMUPLC超高压液相色谱、Xevo TQ 三重四极杆质谱仪,配有电喷雾离子源(美国Waters公司);BS 224 S分析天平(0.1 mg~220 g,德国Sartorius公司);KQ-500DE数控超声波清洗器(昆山市超声仪器有限公司);Multi Reax涡旋混匀仪(德国Heidolph公司);14 000 r/min冷冻离心机(3-30ks,德国Sigma公司);氮吹仪(TTL-DCII型,上海德兆仪器仪表有限公司);HLB固相萃取柱(3 cc,60 mg,美国 Waters公司).
1.2 试剂
氯霉素标准物质(纯度≥99.8%, 德国 Dr.Ehrenstorfer 公司);氯霉素-D5同位素内标(纯度≥95%,德国Dr.Ehrenstorfer 公司);甲醇、乙腈(色谱纯,美国Fisher Scientific公司),乙酸乙酯(色谱纯,美国Dikma公司);甲酸、氨水(色谱纯,美国MREDA公司);氯化钠、无水硫酸钠(分析纯,西亚化学科技(山东)有限公司);盐酸(分析纯,国药集团化学试剂有限公司);纯水处理终端机(Milli Q,美国Millipore公司).
1.3 标准溶液配制及标准曲线绘制
准确称取氯霉素及氯霉素-D5内标,用甲醇作为溶剂,配制1 mg/L的标准储备液,避光保存于-20 ℃冰箱.用10%甲醇水分别将氯霉素和氯霉素-D5内标标准储备液稀释为100 μg/L标准使用液,准确移取氯霉素标准使用液2、5、10、50、100、200、500、1 000 μL至10 mL容量瓶中,加入200 μL同位素内标使用液,用10%甲醇水定容至刻度线,配制浓度范围0.02、0.05、0.1、0.5、1.0、2.0、5.0、10.0 μg/L标准系列,临用现配.
1.4 样品前处理
1.4.1 样品的采集 鸡蛋样品随机采购于北京市各区县商超、农贸市场等,3年共计采购170批次(表1).

表1 2016~2020 年北京市售鸡蛋中氯霉素监测情况
1.4.2 样品的提取与净化 根据氯霉素在碱性条件下可被乙酸乙酯萃取的特点,本研究同时比对了液液萃取法(LLE)和固相萃取法(SPE)的提取净化效率.
SPE法:将鸡蛋破壳后混匀蛋液,准确称取5.00 g(精确到0.01 g)待测液体试样于50.0 mL离心管中,加入100 μg/L同位素内标工作液40 μL.加入5 mL 4% NaCl溶液和5 mL乙腈,涡旋混匀,超声提取5 min,加入10 mL乙酸乙酯,涡旋混匀,以10 000 r/min离心2 min,取上层有机相,待固相萃取净化.
HLB固相萃取柱依次用5 mL甲醇、5 mL 水活化平衡.加入5 mL样品有机相提取液至HLB柱内,待样品过柱后,用5 mL水淋洗,用5 mL甲醇洗脱,收集洗脱液,在50 ℃下用氮气吹干,再加入1 mL 10%甲醇/水溶液混匀超声 1 min,10 000 r/min离心后上清液过 0.22 μm滤膜,用UPLC-MS/MS 分析氯霉素.
LLE法:在待测样液中加入5 g无水硫酸钠后,用15 mL乙酸乙酯,0.45 mL氢氧化铵,反复提取3次,合并上清液,定容后取一定量用正己烷去除脂类,氮气吹干,残留物用1 mL 10%甲醇/水溶液混匀超声1 min,10 000 r/min离心后上清液过0.22 μm滤膜,供仪器检测.
1.5 色谱和质谱检测条件
1.5.1 色谱条件 Waters BEH C18色谱柱(2.1 mm×100 mm,1.8 μm),柱温35 ℃,进样体积为5 μL,梯度洗脱,流动相A为水和0.1%甲酸水(V/V)进行比对,流动相B分别选择了乙腈、甲醇进行比对,流速为0.25 mL/min.洗脱程序为:0~0.5 min,90%~90% A;0.5~2.0 min,90%~10% A;2.0~3.0 min,10%~90% A;3.0~3.5 min,10%~90%A;3.5~5.0 min,90%A.
1.5.2 质谱条件 电喷雾离子源;ESI-模式;毛细管电压0.5 kv,离子源温度150 ℃,脱溶剂温度400 ℃.氯霉素定性定量离子对及相关数据见表2.

表2 液相色谱-串联质谱分析参数
1.6 添加回收实验
对24份空白鸡蛋样本,分别在定量限,4倍定量限,10倍定量限,20倍定量限,40倍定量限和50倍定量限6个浓度水平开展加标回收及精密度试验,每个浓度4个重复.
1.7 数据处理方法
样品中的氯霉素含量用式(1)计算得出,其中X为样品中氯霉素残留量(μg/kg);V为测试液定容体积(mL);m为测试液所代表的样品质量(g),A为样品溶液中氯霉素的峰面积;Ai为样品溶液中氯霉素内标物的峰面积;Ci为样品溶液中氯霉素内标的浓度(μg/L).
(1)
1.8 居民鸡蛋中氯霉素膳食暴露风险评估方法
1.8.1 风险评估数据来源 鸡蛋中氯霉素残留量(μg /kg)采用试验测定数据;目标人群平均体质量(kg)采用2014年北京市国民体质监测结果公报[13]中发布的各年龄及性别人群的体质量,见表4;鸡蛋消费数据来源于北京地区居民膳食调查[14].
1.8.2 膳食暴露量评估 通过食用被污染的食物是人群暴露的主要途径,本试验以鸡蛋为单一的氯霉素暴露途径,以安全指数[15]进行慢性风险评估,安全指数IAPSC依照式(2)计算;EDIC为危害物日摄入量(μg/kg).本着风险最大化原则,本研究采用试验测得的氯霉素残留最大量与北京地区居民每日鸡蛋摄入量的乘积作为危害物日摄入量,进行测评;SIc为安全摄入量(μg/kg)),根据不同的危害物可采用ADI、PTWI、RDI或RfD等数据;f为校正因子,如果安全摄入量采用ADI、RDI或RfD等以日摄入量为基础的数据时f=1,如果安全摄入量采用PTWI等周摄入量为基础的数据时f=7.mB为体质量(kg).当IAPSC远小于1时,安全;IAPSC小于等于1时,风险可接受;当IAPSC大于1时,应当进入风险管理的预警程序中.
(2)
2 结果与分析
2.1 质谱条件的优化
采用Waters-三重四极杆串联质谱仪(ESI 源) ,在多反应监测(MRM) 模式下对氯霉素(相对分子质量为 323.14)的质谱条件进行选择优化.在负离子模式下,氯霉素进入一级质谱后,产生稳定的[M-H]-分子离子峰,得到 m/z 为321.16的母离子,符合含有两个Cl原子9∶6∶1的丰度比特点.氯霉素二级质谱的特征碎片m/z 为 151.89和256.90(图2),321.16/151.89碎片离子强度更高,因此将其作为定量离子,二者丰度比为2∶1.
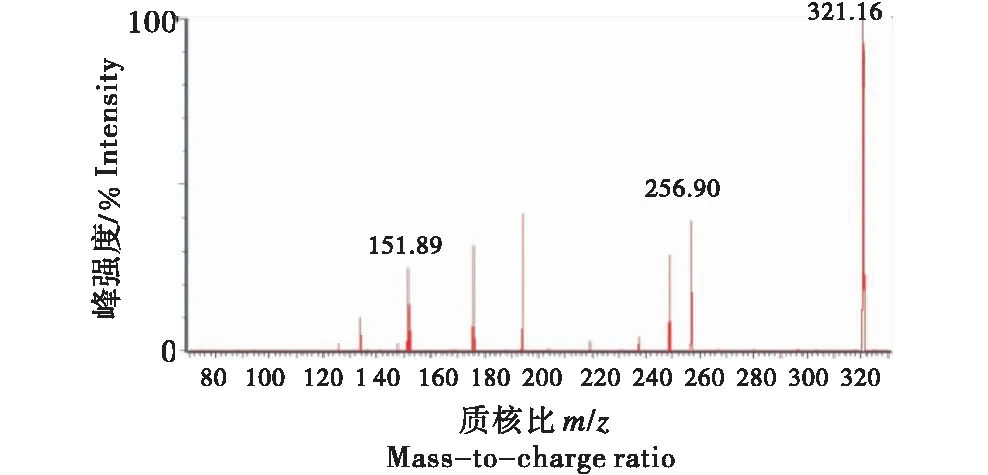
图2 氯霉素质谱图
2.2 色谱条件的优化
分别比较了乙腈和甲醇2种流动相条件,并加入甲酸改性,进行梯度洗脱时氯霉素的响应及分离度.实验结果表明,以乙腈为有机相,氯霉素的色谱峰有明显的拖尾;以甲醇为有机相时得到色谱峰峰形更为对称、峰宽更窄;在有机相和水相中同时添加 0.1%甲酸可以使色谱峰峰形更为尖锐,且响应强度更高,见图3.
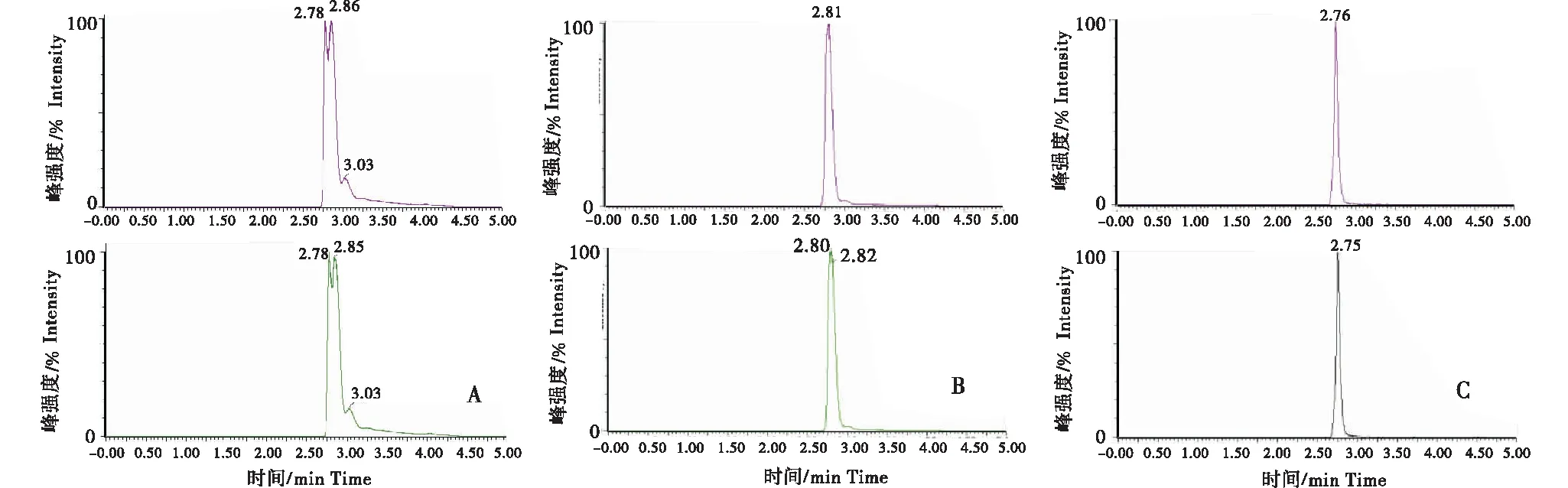
A:乙腈-水;B:0.1%甲酸乙腈-0.1%甲酸水;C:0.1%甲酸甲醇-0.1%甲酸水.
2.3 前处理条件的优化
鸡蛋的基质中含有大量的蛋白质和脂类,且呈现液体状态,待测物与蛋白质相结合溶于蛋液中,难于解离,提取效率低,质谱分析时基质效应严重,净化条件复杂,然而过于繁琐的净化过程也会造成目标待测物提取效率的降低,影响方法的准确性.
分别应用两种方法选取线性范围中段浓度,10倍定量限和15倍定量限,开展加标回收试验,结果见图4.经过对照试验,固相萃取法试验回收率更好,步骤更为简单,使用更少的溶剂,对环境的污染更小,故本研究最终采用固相萃取法进行样品前处理.
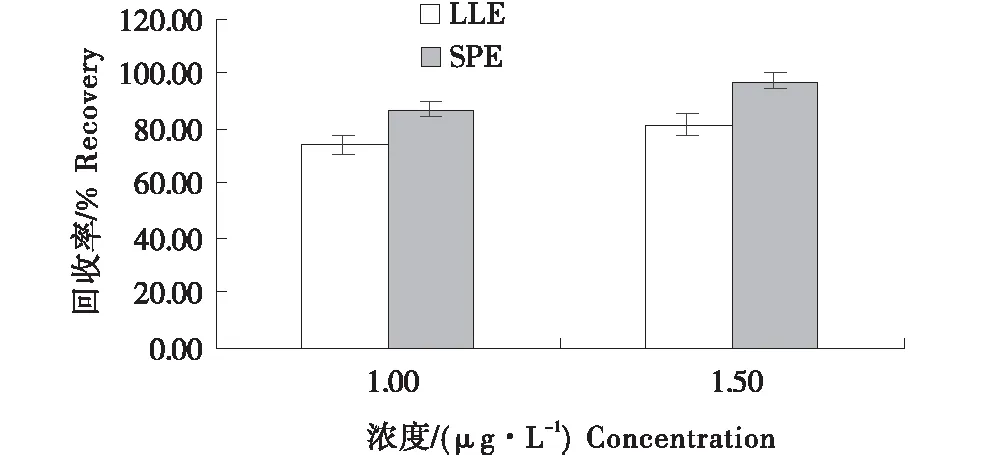
图4 不同净化方式对鸡蛋中氯霉素回收率的影响
试验采用了空白提取液加入标准的方法,比较了两种方法的基质效应.分别测定2种方法鸡蛋空白基质提取液加标1.0 μg/L与同浓度纯溶剂中的响应值,通过两者的比值评价基质效应(ME).ME<0.8说明基质对于待测物有很强的抑制作用;0.8
2.4 线性关系、回收率、精密度及质量控制
以氯霉素与氯霉素内标峰面积比值与内标浓度乘积为纵坐标,质量浓度为横坐标绘制标准曲线.计算公式见式(3),其中A、Ai、C、Ci同式(1).根据式(3)求得标准曲线a和b,以及相关系数r.标准系列溶液按照浓度由低到高依次进样检测,在0.1~5 μg/L范围内有较好的线性关系,线性方程为Y=854.44X-62.86,相关系数r为0.999 5,检出限为0.008 μg/kg,定量限为0.02 μg/kg.对鸡蛋样本添加6种浓度水平,进行加标回收试验,每种进行4个水平测定,结果见表3所示.
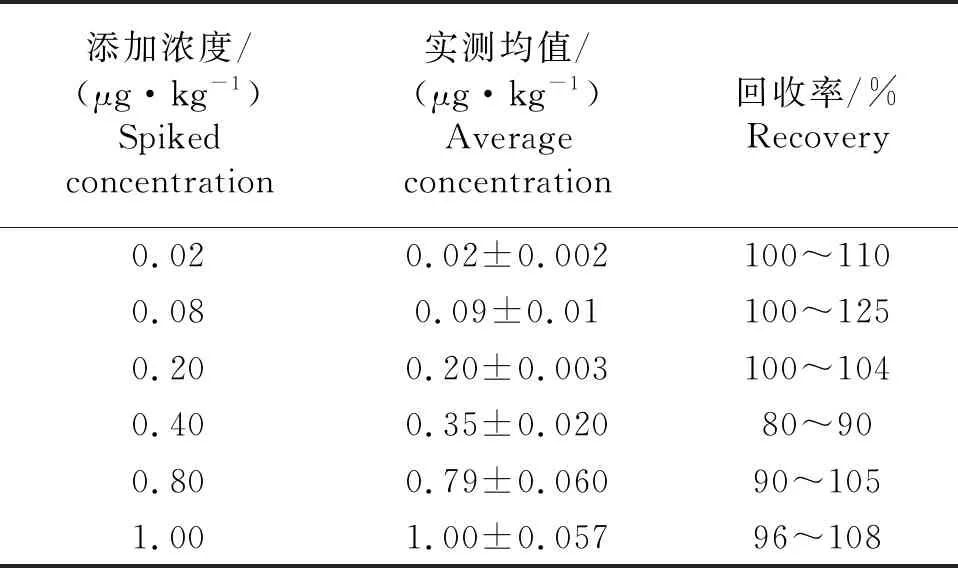
表3 氯霉素的回收率和标准偏差
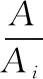
(3)
2.5 不同人群的膳食风险评价
根据北京地区居民膳食调查[14],鸡蛋的日均摄入量为39.3 g/d,由于蛋壳作为非可食用部分,在鸡蛋质量中占比较少,故忽略不计,因此,EDIC值为0.055 μg/d(试验获得鸡蛋中氯霉素残留最大值1.4 μg/kg与0.039 3 kg/d的乘积).世界卫生组织以及联合国粮农组织[16-17]未规定氯霉素安全摄入量限值(JECFA 和JMPR认为添加剂的估计摄入量远远低于任何以常规方法确定的数值),同样,我国也没有对氯霉素安全摄入量做出规定;欧洲食品安全局[18]公布欧洲各国氯霉素慢性膳食暴露限值为幼童11~17 ng/(kg·d),成人2.2~ 4.0 ng/(kg·d),本着风险最大化原则,SIC选用低限进行研究,幼童11 ng/(kg·d),成人2.2 ng/(kg·d).安全摄入量采用日均值,因此校正因子f为1.代入式(2),计算得出北京地区不同年龄段,不同性别居民针对鸡蛋中氯霉素膳食暴露风险安全指数见表4.
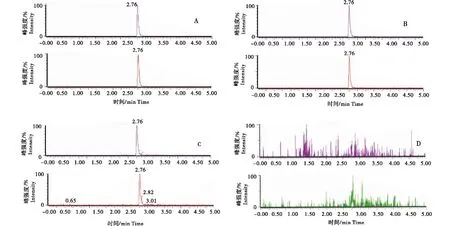
A:2 μg/L标准溶液;B: 2 μg/L空白加标样品;C:1 μg/L阳性样品;D:空白样品.
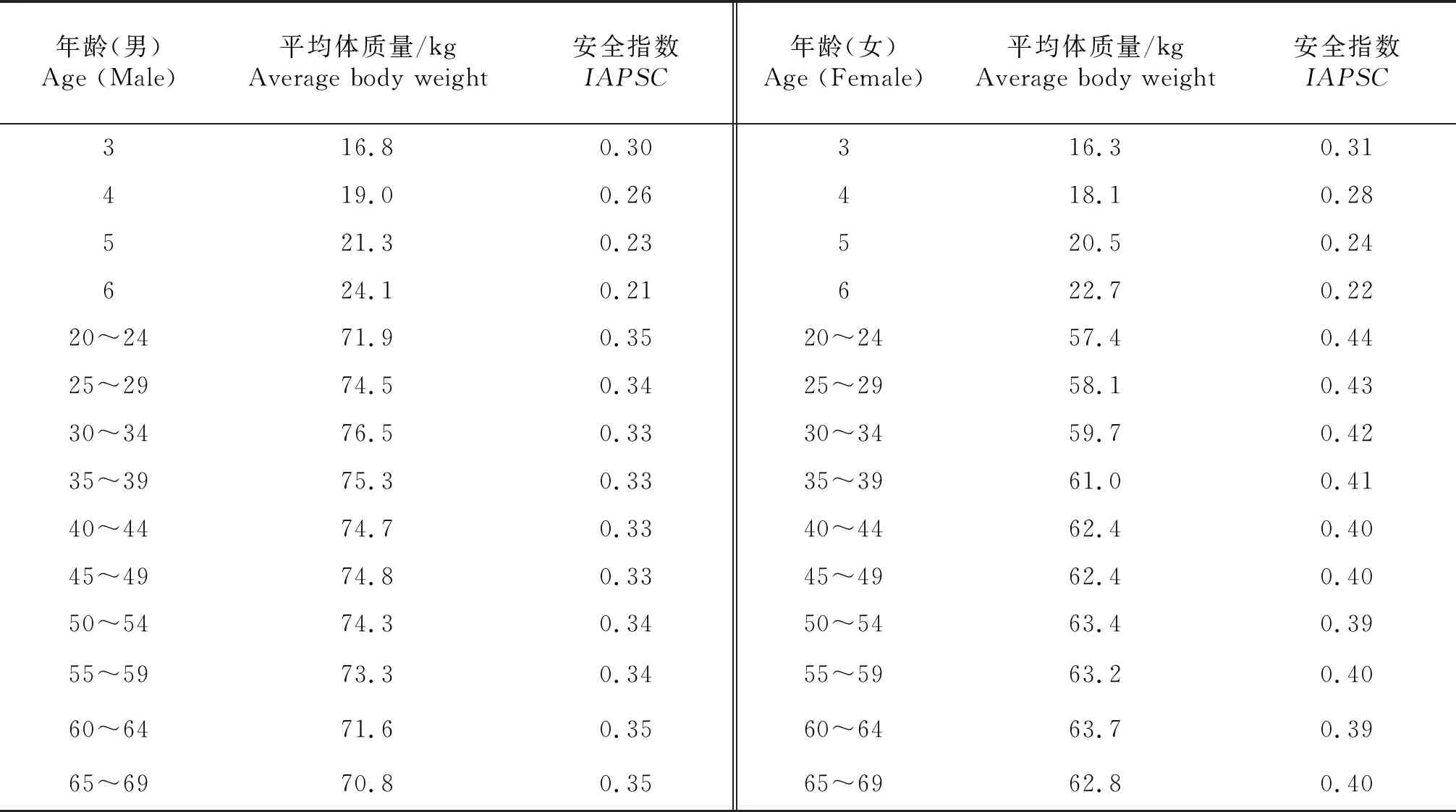
表4 氯霉素在鸡蛋中的膳食风险评估结果
3 结论
本文建立了固相萃取-超高效液相色谱-串联质谱法测定鸡蛋中氯霉素的方法,同位素内标法定量,降低了质谱分析过程中基质效应对检测结果的影响,无内源性干扰.方法稳定性好,准确性高,回收率比较理想,完全可以满足目前风险监测工作检测要求.通过对2016、2018和2020年北京市市售鸡蛋的监测,鸡蛋中氯霉素在居民膳食暴露中风险较低,对3~6岁和20~69岁不同人群膳食暴露的风险均在可接受范围内.
猜你喜欢
现代仪器与医疗(2022年1期)2022-04-19
口腔护理用品工业(2021年4期)2021-11-02
现代仪器与医疗(2021年2期)2021-07-21
分析化学(2019年3期)2019-03-30
分析化学(2019年3期)2019-03-30
中国科技纵横(2019年23期)2019-02-14
科技视界(2018年31期)2018-03-30
分析化学(2018年12期)2018-01-22
海峡科技与产业(2017年1期)2017-03-04
家庭科学·新健康(2016年5期)2016-05-12
