
多条件多样本RNA-Seq 数据的剪切异构体表达水平估计
2021-11-27 00:48张礼马越吴东洋
智能系统学报 2021年6期
张礼,马越,吴东洋
(1.南京林业大学 信息科学技术学院,江苏 南京 210016;2.江苏健康卫生职业学院 中西医结合学院,江苏 南京 210018)
选择性剪切事件是导致生物体多样性的重要原因之一。为了进一步揭示选择性剪切的内在机制,迫切需要计算剪切异构体的表达水平。与传统的基因芯片技术相比,高通量RNA 测序(RNA sequencing,RNA-Seq)技术具有高通量、高灵敏度、可重复性好等优势,已成为转录组学分析的一个标准技术手段[1-5]。
RNA-Seq 测序实验获得海量读段,将读段与参考注释序列进行匹配,之后便可估计剪切异构体的表达水平。但是在估计剪切异构体表达水平的过程中,面临着两个最大挑战,即读段的多源映射和数据偏差[6-7]。研究者提出了大量剪切异构体表达水平估计方法来解决上述的问题。rSeq方法把读段映射到外显子的过程当作一个泊松随机过程,其泊松分布的参数对应着基因所包含剪切异构体表达水平的线性加权[8]。但是rSeq 方法假设基因上读段分布是均匀的,这与真实数据分布特点不一致。在真实数据中,读段分布呈现明显的非均匀特征。读段的非均匀分布主要是由测序数据中的各种偏差造成的,比如GC 碱基序列偏差,5 端和3 端的位置偏差以及实验技术性偏差等。针对偏差所导致问题,NURD 方法考虑了全局和局部位置偏差所带来的影响[9]。POME 方法考虑了序列中碱基之间的关联性[10]。为了考虑更复杂的偏差,大量概率生成式模型被提出,其直接模拟读段的随机采样过程。Cufflinks 方法设计了不同的模型来消除序列偏差和位置偏差的影响,从而更加准确地描述读段随机采样过程[11]。BitSeq 和PBSeq 方法采用了与Cufflinks 同样的偏差估计模型[12-13]。RSEM 方法考虑了读段匹配的不确定性因素,并且使用了读段起始位置的经验分布来表示读段在基因上的非均匀分布特征,但是其未考虑序列偏差这个重要因素[14]。上述方法采用不同的偏差估计模型来模拟读段的非均匀分布特征,都能提高剪切异构体表达水平的估计准确程度。
由于数据噪声和偏差的影响,异构体表达水平的准确性仍然有较大提高的空间[15-16]。常规的RNA-Seq 测序实验通常会设置不同的实验条件,比如:同一个细胞组织下参照组和对照组,不同时间点下胚胎发育状况等。此外为了避免实验中的技术性误差,同一个实验条件下会进行多次重复性技术性实验。这使得一次测序实验获得的RNA-Seq 数据集是一个多条件多样本的数据集。但是上述方法都是假设RNA-Seq 数据集中各个样本之间是相互独立,因此都是单独逐个处理每个数据样本。这导致样本之间的相关性没有得到充分利用。因此有少量工作开始探索联合多样本RNA-Seq 数据进行异构体表达水平估计[17-18]。Sequgio 方法能从多样本数据中自动获取位置偏差和局部序列影响,再通过对联合统计模型添加一个光滑的正则化项,来控制读段在多样本的一致性[19]。MSIQ 方法考虑多样本之间的异质性所导致的结果不稳定性,首先将同质性相近的样本归为同一组,然后在贝叶斯框架模型下,给同一组之内的样本赋予较高的权重,从而获得更加鲁棒的异构体表达水平[20]。XAEM 方法采用双线性模型同时估计异构体表达水平和数据偏差,该模型能够自动对潜在的未知偏差进行经验校正[21]。但是上述方法所处理的多样本数据,仅仅是针对单条件下的多样本,比如同一个组织细胞的对照组或者同一个时间点状态。当处理多条件多样本数据时,这些方法都是假设各个条件之间不相关,把多条件多样本数据拆分为多个单条件多样本数据集来进行异构体表达水平计算。但是基因读段分布在不同条件下同样具有高度相似性[22]。为了充分利用数据信息,PGSeq 方法采用泊松分布和伽玛分布的混合模型联合估计基因和异构体表达水平,其伽玛分布用来模拟基因读段分布在多条件多样本下的偏差信息[23]。但PGSeq 方法未考虑到基因和异构体表达水平之间的稀疏特性,易受到数据噪声的影响。
基于上述问题,本文提出了一个多条件多样本RNA-Seq 测序数据异构体表达水平估计方法,MCMS-Seq(multi-condition multi-sample RNASeq)。该模型考虑了基因读段分布在不同条件下的样本具有高度相似性,设计一个联合多条件多样本数据的偏差估计模型,同时考虑了基因读段分布受全局偏差和局部偏差的影响。此外,MCMS-Seq方法增加了L2/L1组稀疏约束和L1稀疏约束两个正则化项,用来体现基因和剪切异构体之间存在稀疏特性,以及消除技术性误差和数据噪声的影响。最后,通过3 个多条件多样本RNA-Seq 数据集来评估MCMS-Seq 方法的性能。
1 MCMS-Seq 方法
1.1 MCMS-Seq 模型表示
由于选择性剪切事件在真核生物中普遍存在,这给计算剪切异构体表达水平带来了一个最大问题,即如何定量确定匹配到共享外显子上的读段来自哪个剪接异构体。图1 中显示的基因包含4 个外显子(Exon)和3 个剪切异构体。其中一个外显子可以同时被多个剪切异构体共享,比如外显子1 被3 个剪切异构体共享,但是剪切异构体2 仅共享了外显子1 的部分序列。针对这类部分共享情况,可将外显子1 分割为2 个不重叠的外显子片段。因此该基因的4 个外显子被分割成7 个完全不重叠的外显子片段。映射矩阵A表示图1 中剪切异构体与外显子片段的关系,其中矩阵元素a12=1 表示异构体1 包含外显子片段2。当测序读段匹配到基因上,外显子片段上的读段数目即可被统计出来。假设某个数据集有2 个条件每个条件包括2 个样本,总计4 个样本,那么图1 中基因在不同样本中读段数据可用数据矩阵D表示。每一行表示该基因在一个样本中外显子片段的读段数目。

图1 剪切异构体中外显子片段划分示例Fig.1 Example of exon segmentation in an isoform
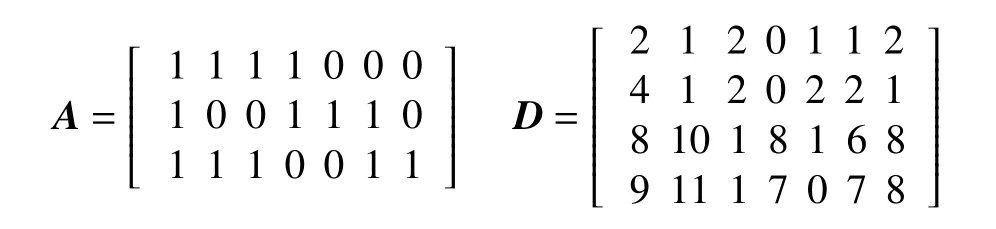
假设测序实验获得RNA-Seq 数据包含C个条件,每个条件包含N个样本。对于基因g,该基因包含K个剪切异构体和M个外显子片段,其与外显子片段的映射关系由映射矩阵AM×K表示。yci j表示基因第j个外显子片段在第c个条件中第i个样本上的读段数目。根据实验原理,基因外显子片段的读段数目等于共享该外显子片段的剪切异构体上读段之和,其数学模型为

式中:xcik表示基因第k个剪切异构体在第c个条件中第i个样本;ajk表示剪切异构体与外显子片段之间的映射关系;wci表示第c个条件中第i个样本的读段总数;lj是第j个外显子片段的长度。
式(1)模型是基于基因读段是均匀分布假设的前提,但是实际数据中,基因读段分布呈现明显的非均匀特征。由于基因读段分布模式在不同条件不同样本下具有高度相似性,因此假设bj表示第j个外显子片段的偏差权重,其值在样本之间是共享的。现将偏差bj融入到式(1)中,得到如下模型:

对于多条件多样本的RNA-Seq 数据集,基因g所包含的K个剪切异构体的表达水平X可以通过回归模型计算,其公式如下:

所有剪切异构体在不同样本中的表达水平都要求xcjk≥0。为了便于理解和计算,式(3)可以简化为矩阵形式:

式中D表示归一化后的数据矩阵。
一个基因虽然包含多个剪切异构体,但是在不同条件下,少数剪切异构体的表达水平决定了该基因的表达。因此基因和剪切异构体表达水平之间具有稀疏特性。通过对剪切异构体表达水平X增加L1范数来保留稀疏特性,式(4)可改写为

虽然模型增加了L1范数的稀疏约束,但仍然会发现出现大量低表达的剪切异构体,而这部分剪切异构体不全是真实的低表达。当一个剪切异构体在同一个条件下的所有重复样本都是低表达水平,那么可认为此剪切异构体是真实的低表达。而对于零散出现的低表达剪切异构体,则受到数据噪声和偏差的影响,不是真实的低表达。为了消除虚假的低表达剪切异构体的影响,在式(5)的基础上增加了L2/L1组稀疏约束得到了MCMSSeq 方法的最终形式:

式中 λ1和 λ2分别是L2/L1和L1约束的系数。通过两个稀疏约束项,MCMS-Seq 方法不仅考虑了基因和剪切异构体表达水平之间的稀疏性质,同时也可以消除数据噪声和偏差对低表达剪切异构体的影响。图2 显示了MCMS-Seq 方法的优化问题。

图2 MCMS-Seq 方法的优化问题Fig.2 Optimization problem of MCMS-Seq method
1.2 多条件多样本偏差估计模型
在多条件多样本数据中,图3 显示了基因的读段分布无论在不同条件下,还是在同一个条件的重复样本中,其分布模式具有高度相似性。MCMS-Seq方法提出了一个基于多条件多样本的读段非均匀偏差估计模型。该偏差估计模型由两部分构成:全局偏差 βglobal和局部偏差 βlocal。全局偏差 βglobal的读段非均匀分布模式是从数据集中所有表达基因中获得。由于读段多源映射会影响基因读段分布,全局偏差估计仅仅选择只包含单个剪切异构体的基因。此外,由于低表达水平基因的不确定性,读段计数小于50 的基因被排除。将筛选后的基因均分为20 个等长度的区间,统计并归一化每个区间内读段数目。最后采用多项式回归来拟合基因每个区间上的读段数目,得到的拟合曲线表示基因读段分布的全局偏差特征。而局部偏差βlocal仅仅统计基因每个外显子片段在多条件多样本数据上的读段数目,再进行均一化处理,其反映了单个基因自身的读段分布特征。

图3 小鼠数据集中基因Utrn 读段分布Fig.3 Read distributions of gene Utrn in the mouse dataset
一旦获得数据集的全局偏差曲线和单个基因的局部偏差特性,便可以计算出基因上每个外显子片段的偏差值:
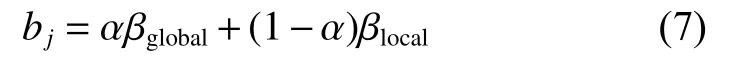
式中:α 是权重参数,用来权衡全局偏差和局部偏差的影响。本文选择α=0.5,表示全局偏差和局部偏差对基因读段分布具有相同的影响[9],不仅仅能反映读段非均匀分布在多条件多样本之间具有高度相似的特征,同时还可以体现出每个基因独有读段分布特点。
1.3 MCMS-Seq 模型实现
MCMS-Seq 方法的实现可以分为3 个部分:读段数据预处理、基因偏差估计和表达水平估计。
1) 读段数据预处理,是从匹配成功的读段数据中统计基因每个外显子片段的读段计数,以及从注释文件中获得外显子片段和剪切异构体之间的映射关系矩阵。
2) 基因偏差估计,是计算数据集的全局偏差和基因的局部偏差,从而获得基因每个外显子片段的基因偏差值。
3) 剪切异构体表达水平估计,由于模型是针对多条件多样本数据集,同时模型包含L2/L1和L1约束,MCMS-Seq 方法采用SPAMS 优化工具箱来求解[24-25]。
MCMS-Seq 方法的详细流程如算法1 所示,采用Python 和MATLAB 混合编程实现。
算法1MCMS-Seq 方法
输入多条件多样本数据,注释文件;
输出每个基因的剪切异构体表达水平。
1)数据预处理:统计外显子片段读段数目矩阵D,构建映射关系矩阵A。
2)基因偏差估计:计算外显子片段偏差值。
3)表达水平估计:计算所有基因的X∗。
1.4 多条件多样本数据分析通道
为了方便用户使用MCMS-Seq 方法,本文提供了一个多条件多样本RNA-Seq 测序数据分析通道,如图4 所示。当获得RNA-Seq 测序数据样本后,使用经典读段匹配软件Bowtie[26],将每个数据样本的读段匹配到参考转录组参考序列上。每个样本匹配结果作为输入数据一并输入到MCMSSeq 分析通道中,从而可获得剪切异构体在不同样本中的表达水平。一旦获得剪切异构体的表达水平,可提供给高层次的后续分析使用。

图4 多条件多样本RNA-Seq 数据分析通道Fig.4 Analysis pipeline of multi-condition multi-sample RNA-Seq data
2 实验结果与分析
本文选择了经典方法Cufflinks(v.2.2.1)和PGSeq(v.1.0),以及最新方法XAEM(v.0.1.1),分别在 3个数据集上与MCMS-Seq 方法进行比较,用来验证剪切异构体表达水平的性能。针对多条件多样本数据集,Cufflinks 是每个样本单独处理,而PGSeq、XAEM 和MCMS-Seq 都是多个样本联合处理。
2.1 数据集
3 个多条件多样本的RNA-Seq 数据集被用来验证MCMS-Seq 方法估计剪切异构体表达水平的准确性。3 个数据集分别是小鼠数据集、人类大脑的SEQC 和MAQC-II 数据集,它们都来自Illumina/solexa 测序平台。
小鼠数据集包含3 个条件,分别是肝脏、大脑和骨骼肌3 个组织,其中每个组织分别包含了 2个重复实验样本。使用 RefSeq 数据库的基因注释信息(GRCm38/mm10),总共包含 33608 个剪切异构体,主要用来验证同条件下重复样本之间剪切异构体表达水平的可重复性[27]。
MAQC(micorarray quality control)来自美国药品监管局的生物芯片质量控制项目。该项目分为三期实施,即MAQC-I、MAQC-II 和MAQC-III,其产生的数据集被广泛应用于评估不同测序平台下不同方法的性能。本文主要利用了MAQC-II 和MAQC-III 两期项目提供的数据。MAQC-III 也被称为SEQC(sequencing quality control)。SEQC 包括两个实验条件UHRR(universal human reference rna)和HBRR(human brain reference RNA),每个条件分别有8 个重复实验样本。SEQC 数据集提供了两万多个经qRT-PCR 实验验证的剪切异构体。与Ensembl 注释信息(GRCh37/hg19)相匹配后,最终得到16603 个剪切异构体。这些剪切异构体的qRT-PCR 值被当作真实表达水平值,可用来评估模型计算剪切异构体表达水平的准确性[28]。
基因表达水平是由其包含的剪切异构体所构成,因此基因表达水平可用来进一步验证剪切异构体表达水平的准确性。MAQC-II 数据集同样包含UHRR 和HBRR 两个实验条件,每个条件下包含7 个重复性实验。该数据提供了1000 个经qRT-PCR 实验验证的基因。根据与Ensembl 注释信息(GRCh37/hg19)相匹配,最终获得838 个基因。这些基因的qRT-PCR 值被当作真实基因表达水平值,用来间接评估模型计算剪切异构体表达水平的准确程度[29]。
2.2 多条件多样本偏差估计模型验证
MCMS-Seq 方法提出了一个基于多条件多样本偏差估计模型,同时考虑了读段分布受到全局偏差和局部偏差的影响,用来获取读段分布在样本之间的高度相似性特征。SEQC 数据集被用来验证偏差估计模型的有效性。图5 显示使用该模型对SEQC 数据集的偏差估计流程。从图5(a)中可以看出,在SEQC 数据集中,基因的读段分布呈现明显的非均匀分布特征,特别是在基因的两端。这个现象符合基因的 3′端和 5′端最容易受到RNA-Seq 测序技术影响的事实。选择基因Cdca4来展示估计全局偏差和局部偏差的过程。基因Cdca4 包含3 个剪切异构体和5 个外显子片段,其结构如图5(b) 所示。图5(c)是通过多项式回归拟合图5(a)读段分布所得到SEQC 数据集全局偏差曲线。曲线上黑点表示基因Cdca4 外显子片段长度的比率。通过长度比率在曲线上的取值,可得到Cdca4 基因中每个外显子片段的全局偏差值。统计并归一化基因Cdca4 的外显子片段在所有样本中的读段数目,即可获得该基因的局部偏差,如图5(d) 所示。从图5 中可以看出,该基因在3′端和 5′端受到的局部偏差影响要略小于全局偏差。为了进一步验证基因的局部偏差,从SEQC 数据集中随机选择4 个基因:Plagl1、Eif4a、Sv2b 和Whrn,其分别包含5、6、7、8 个剪切异构体。从图6 中可以看出,不同基因的局部偏差整体上都呈现明显非均匀分布特征,但是单个基因之间存在一定差异,比如基因Whrn 中间外显子的偏差值表现出由高到低的趋势。因此,MCMSSeq 方法提出的多条件多样本偏差估计模型,不仅能反映在多条件多样本数据中读段非均匀分布具有高度相似性的特征,同时还可以体现出单个基因独有读段分布特点。

图5 MCMS-Seq 方法的偏差估计流程Fig.5 Bias estimation process of the MCMS-Seq method

图6 基因的局部偏差分布Fig.6 Local bias distribution of genes
2.3 多条件多样本下剪切异构体表达水平的验证
MCMS-Seq 方法处理多条件多样本数据集时是联合所有样本同时处理,通过增加稀疏约束,不仅可以消除数据噪声的影响,同时也能体现基因和剪切异构体之间存在的稀疏特性。选择小鼠数据集的基因Nph2 来验证,该基因包含3 个剪切异构体。
在小鼠数据集中,同一个剪切异构体在同一个条件下的多个重复样本中,其表达水平应该是相近的。若一个剪切异构体在重复样本中零散地出现低表达,则此剪切异构体的表达水平受到数据噪声的影响。传统方法Cufflinks 都是每个样本依次单独处理,其表达水平值如表1 所示。NM_001 364736 表达水平在Muscle 条件两个重复样本中就可能受到数据噪声的影响,NM_157294 在Liver 条件下也存在同样的情况。表2 中XAEM 方法获得的NM_001364736 和NM_157294 表达水平都是极低值,极大可能是受到数据噪声的干扰。MCMS-Seq 方法联合处理多条件多样本数据集。从表3 中可以看出,NM_001364736 在3 个组织条件下都未表达,NM_157294 在大脑和骨骼肌组织条件下具有真实的低表达,而在肝脏组织条件下未表达,能有效消除数据噪声的影响。

表1 Cufflinks 估计基因Nhp2 中3 个剪切异构体表达水平Table 1 Expression level of three isoforms in Nph2 gene estimated using cufflinks

表2 XAEM 估计基因Nhp2 中3 个剪切异构体表达水平Table 2 Expression level of three isoforms in Nph2 gene estimated using XAEM
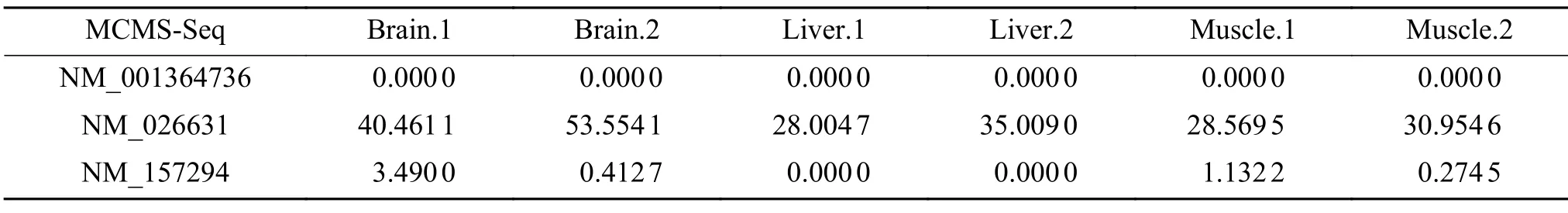
表3 MCMS-Seq 估计基因Nhp2 中3 个剪切异构体表达水平Table 3 Expression level of three isoforms in Nph2 gene estimated using MCMS-Seq
此外,基因外在表现通常是由其包含的少数剪切异构体决定的,因此基因和剪切异构体之间存在稀疏特性。在表4 中,PGSeq 方法得到的3 个剪切异构体表达水平都存在较高的表达值,无法体现稀疏特性。而Cufflinks 和XAEM 受数据噪声影响,同样很难体现出该数据特性。MCMSSeq 方法增加了L2/L1组稀疏和L1稀疏约束来考虑上述生物特性。NM_026631 在所有组织条件下中都有较高的表达水平,说明基因Nph2 的表达主要由NM_02663 所决定。NM_001364736 在3 个组织条件下都未表达,特别在肝脏组织条件下NM_157294 和NM_001364736 同时未表达,这表明基因Nph2 在肝脏中只有剪切异构体NM_026631 参与基因表达。因此,MCMS-Seq 方法能体现基因表达是由少数剪切异构体所决定的生物特性,提供了更好的生物可解释性。

表4 PGSeq 估计基因Nhp2 中3 个剪切异构体表达水平Table 4 Expression level of three isoforms in Nph2 gene estimated using PGSeq
2.4 剪切异构体的可重复性验证
在多条件多样本测序实验中,同一个条件下设计多重复性样本是为了避免技术性误差所带来的影响。这使得同一个剪切异构体在同一个条件下的重复样本之间的表达水平是相近的。小鼠数据集被用来验证剪切异构体表达水平在样本之间的可重复性。采用Person 相关系数来评估可重复性,其值越高说明能更加有效地消除技术性误差所造成的偏差。由于RNA-Seq 测序技术得到表达水平其幅度跨度很大,Person 相关系数易受到少数高表达的剪切异构体影响。因此在计算相关系数之前,对所有剪切异构体表达水平进行对数转换,从而避免上述问题。表5 中显示不同方法在小鼠数据集上不同条件下的相关系数值。从表中可以看出,MCMS-Seq 方法在肝脏、大脑和骨骼肌3 个条件下都获得了比其他3 个方法更好的结果。尽管MCMS-Seq 方法是面向处理多条件多样本数据集,但仍然可以保证剪切异构体在同一个条件中下样本之间具有高度的可重复性。这也符合RNA-Seq 测序实验中设计重复实验的目的。

表5 在小鼠数据集上不同方法估计的剪切异构体表达水平在样本之间的相关系数Table 5 Correlation coefficients between isoform expression levels estimated using various methods in the mouse dataset
2.5 PCR 剪切异构体的表达水平验证
SEQC 数据集被用来验证不同方法估计剪切异构体表达水平的准确性。该数据集提供了16 603个经过qRT-PCR 验证的剪切异构体,这些剪切异构体被当作基准数据。计算不同方法得到剪切异构体表达水平与qRT-PCR 值之间的相关系数。从表6 中结果可以看出,MCMS-Seq 方法在UHRR条件上稍微优于PGSeq 方法,而在HBRR 条件上获得较为明显的提升。尽管XAEM 方法是多样本处理,但获得最差的性能,其可能是该方法对数据偏差考虑得不够。整体上说,MCMS-Seq 方法估计的剪切异构体表达水平能取得较为准确的结果。

表6 在 SEQC 数据集上不同方法与 qRT-PCR 验证剪切异构体之间的相关系数Table 6 Correlation coefficients between qRT-PCR values and isoform expression levels estimated using various methods in SEQC dataset
2.6 PCR 基因的表达水平验证
现实中包含qRT-PCR 验证的剪切异构体数据集很少,而基因的表达水平是由其所包含的剪切异构体所决定的,因此可以通过验证qRTPCR 验证基因的表达水平来间接验证剪切异构体表达水平的准确性。MAQC-II 数据集被广泛地应用于评估不同方法估计基因表达水平的性能。MAQC-II 数据集提供了838 个qRT-PCR 验证的基因,这些基因总共包含了6927 个剪切异构体。Cufflinks 和PGSeq 方法提供了基因的表达水平,XAEM 和MCMS-Seq 方法的基因表达水平由所对应的剪切异构体表达水平求和得到。表7 显示了不同方法得到的基因表达水平与qRT-PCR 值之间的相关系数。从表7 中可以看出,相比其他方法,MCMS-Seq 方法得到了更好的准确性。

表7 在 MAQC-II 数据集上不同方法与 qRT-PCR 验证基因之间的相关系数Table 7 Correlation coefficients between qRT-PCR values and isoform expression levels estimated using various methods in the MAQC-II dataset
2.7 稀疏参数的选择
MCMS-Seq 方法包含了L2/L1组稀疏约束和L1稀疏约束两个正则化项,不仅用来考虑基因和剪切异构体之间的稀疏特性,同时用来消除虚假低表达剪切异构体带来的影响。在式(6)中,参数 λ1和 λ2分别对应着L2/L1组稀疏约束和L1稀疏约束,其值的选择能影响到剪切异构体表达水平的准确性。当 λ1或 λ2→+∞ 时,都会导致剪切异构体出现不表达情况,区别在于,λ1→+∞ 会导致同一个剪切异构体在不同条件下都没有表达。当λ1→0 时,剪切异构体的表达水平容易受到数据噪声的影响,产生虚假的低表达。而 λ2减小时,基因与剪切异构体之间的稀疏特性将减弱。选择SEQC 数据集中HBRR 条件来分析参数选择对剪切异构体表达水平准确性的影响。假设参数λ1和 λ2分别选择0.1、1、10 和100 这4 个值,图7显示了在取不同参数值时,MCMS-Seq 方法估计的剪切异构体表达水平与qRT-PCR 验证的剪切异构体之间的相关系数。从图7 可以看出,当 λ1和 λ2同时增大时,其相关系数都显著下降,因为有大量真正表达的剪切异构体被估计成未表达。而 λ1和 λ2在取值1 附近能获得较为稳定的结果,因此本文中所有实验都是设定 λ1和 λ2为1。
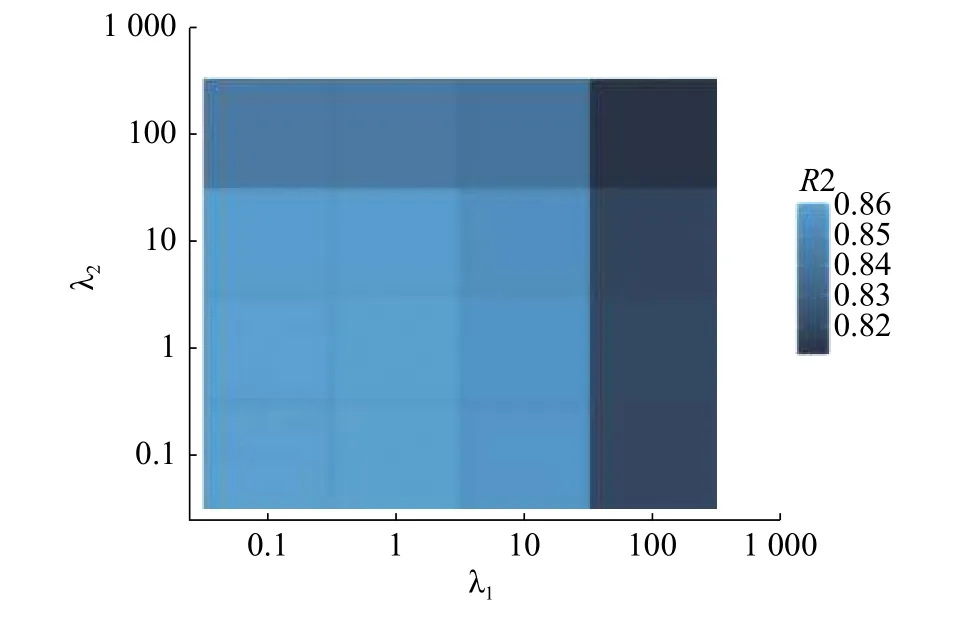
图7 参数 λ1和 λ2 对剪切异构体表达水平的影响Fig.7 Effect of isoform expression levels by parameters λ1and λ2
3 结束语
本文提出了一个基于多条件多样本RNASeq 测序数据的剪切异构体表达水平估计方法。为了考虑基因读段分布在不同条件下的高度相似性,MCMS-Seq 方法设计一个联合多条件多样本的偏差估计模型,同时考虑了基因读段分布的全局偏差和局部偏差所带来的影响。从数据分析可以看出,该偏差估计模型能较为准确地描述出基因读段非均匀分布特性。此外,MCMS-Seq 方法增加了L2/L1组稀疏约束和L1稀疏约束两个正则化项,体现了基因和剪切异构体之间存在稀疏的生物特性,同时消除了技术性误差和数据噪声的影响。在小鼠数据集上,MCMS-Seq 方法估计的剪切异构体表达水平能获得更好的可重复性。通过与SEQC 数据集中qRT-PCR 剪切异构体和MAQC-II 数据中qRT-PCR 基因的验证,MCMSSeq 方法比其他3 个对比方法更佳的性能。
由于大量多条件多样本数据集是时序数据集,蕴含了时间信息,但是MCMS-Seq 模型未考虑到数据中的时间信息。在未来的研究中,可以考虑在模型中融入时间信息,从而进一步提高剪切异构体的表达水平的准确性。此外,可将MCMS-Seq 模型推广到单细胞测序数据分析,可提供更好的生物解释性。
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
电子科技大学学报(2022年5期)2022-10-29
云南化工(2021年8期)2021-12-21
中国生殖健康(2020年4期)2021-01-18
山东冶金(2018年5期)2018-11-22
中国生殖健康(2018年4期)2018-11-06
西安建筑科技大学学报(自然科学版)(2016年1期)2016-11-08
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
铁道科学与工程学报(2015年4期)2015-12-24
