
第六配位基对S=2[FeⅣ(O)(TQA)(L)]n+配合物反应活性的影响
2021-12-22 03:03李清月,李文志,王禹茜,王一,贺晓阳
大连工业大学学报 2021年6期
李 清 月,李 文 志,王 禹 茜,王 一,贺 晓 阳
(1.大连工业大学 生物工程学院,辽宁 大连 116034;2.大连工业大学 信息科学与工程学院,辽宁 大连 116034 )
0 引 言
含有单核非血红素铁活性中心的氧化酶在环境、生理以及药物开发等领域都具有重要的作用。单核非血红素铁氧活化酶催化反应的中间体通常是高价铁氧配合物,与血红素酶一样可以参与许多重要的催化循环过程。迄今为止,生物无机化学家们已经合成90多种四价非血红素铁模型配合物,其中,有13种可以测出晶体结构[1]。在四价非血红素铁模型配合物中,超过90%的配合物的基态为三重态。2003年,Rohde等[2]首次合成可以独立并稳定存在的基态为三重态的六齿配位四价铁氧配合物并解析出晶体结构,随后合成了以N4和N4S为配位基的四价铁氧配合物[3-5],其性质可以由莫斯堡谱、拉曼光谱、X衍射和理论计算等手段检测。

1 计算方法
利用B3LYP泛函(Becke,three-parameter,Lee-Yang-Parr)[10],通过高斯09软件包对[FeⅣ(O)(TQA)]2+、[FeⅣ(O)(TQA)(AN)]2+和[FeⅣ(O)(TQA)(OTf)]+的几何结构进行优化。Fe原子使用double-ζ LACVP基组,C、H、O和N原子使用6-31G**基组优化(简称B1)[11]。在单点能计算中,原子都用TZVP[12-13]基组(简称B2)。优化与反应通道计算都在溶剂里进行,模型为自洽反应场的连续极化模型,溶剂选取乙腈。稳定结构没有虚频,过渡态只有一个虚频,且沿着反应坐标的方向。
利用The Rate程序进行Eckart隧穿效应计算,计算传输系数k,并通过公式计算隧穿效应对能垒的影响。
式中:R为通用气体常数,T为绝对温度。
实验速率通常在273 K收集[14]。
本研究利用Multiwfn程序[15-16]计算了Hirshfeld 电荷和轨道布局。
2 结果与讨论
2.1 [FeⅣ(O)(TQA)(L)]n+(L=None,AN,OTf-)的几何结构和电子结构
[FeⅣ(O)(TQA)(L)]n+(L=None,AN,OTf-)有三个自旋态:单重态、三重态和五重态。配位基的结构决定了三种配合物的基态为五重态。五重态时,Fe上四个d电子的排布:[FeⅣ(O)(TQA)]2+的电子构型为π*xz1π*yz1σ*xy1σ*x2-y21σ*z20,[FeⅣ(O)(TQA)(AN/OTf)]2+/+的电子构型为δ*xy1π*xz1π*yz1σ*x2-y21σ*z20(图1)[3]。
表1列出了三种配合物的Fe原子和O原子Hirshfeld电荷和布局。图2给出了[FeⅣ(O)(TQA)(L)]n+的几何结构图和相对应几何结构参数。如图2所示,五重态时,三种配合物的Fe—O键长分别为0.163 0、0.162 8和0.162 3 nm。Fe和竖直方向的N之间的键长(Fe—Naxial)分别为0.202 1、0.207 3和0.209 3 nm。与相对应的三重态的Fe—Naxial键长相比,五重态的键长更长。六配位的两种配合物的Fe与赤道方向的N的平均距离(Fe—Neq)要长于五配位的配合物。三重态时,五配位的配合物Fe与赤道方向的N的平均距离与五重态时几乎一致,而对于六配位的配合物五重态的键长要长于三重态。在B2//B1+ZPE的计算水平下,三种配合物的基态为五重态,其三重态和五重态的能量差分别为61.95、11.30和22.60 kJ/mol。如图1所示,对于五配位的配合物,dx2-y2和dxy轨道是简并的,所以,三重态的能量要远高于五重态的能量。而对于六配位的配合物,dx2-y2和dxy轨道之间有能量差,所以五配位的配合物[FeⅣ(O)(TQA)]2+三重态和五重态的能量差要远高于六配位的配合物。
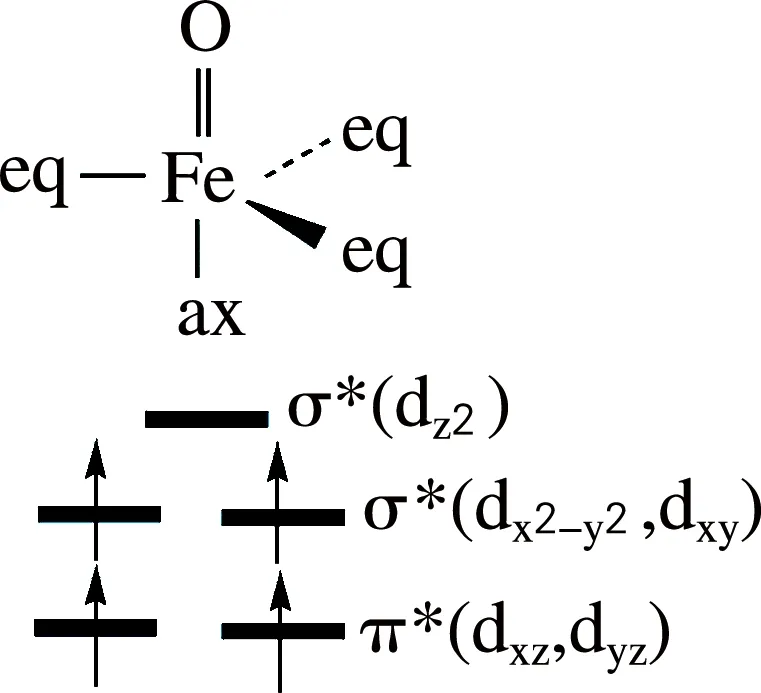
(a)TQA
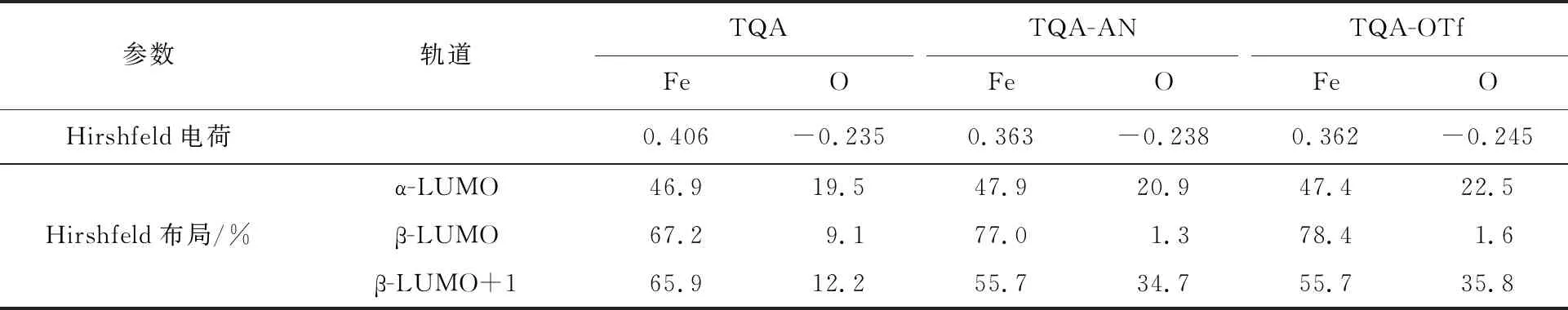
表1 铁原子和氧原子的Hirshfeld电荷和布局Tab.1 Hirshfeld charges and distributions of the Fe and O atoms
2.2 分子内氢转移反应
TQA配体的体积庞大,铁氧中心和配体的氢原子之间存在着空间相互作用,这样的结构有利于形成高自旋态的反应物。为了更好地说明TQA与FeO单元的O之间的关系,计算了O夺取TQA配位基上H的反应能垒。如图3所示,在B2//B1的计算水平下,五重态时反应能垒分别为112.60、113.02和117.04 kJ/mol。由此可见,三种配合物发生分子内的氢转移反应是几乎不可能的,这一性质与第六配位基的性质无关。

(a)TQA
2.3 C—H键的羟基化反应和CC键的环氧化反应
优化了三种配合物催化氧化丙烯反应的几何结构。所有构型优化都是在B1计算水平下进行,单点能的计算在B2计算水平下进行。
2.3.1 C—H键的羟基化反应
(a)5TQA
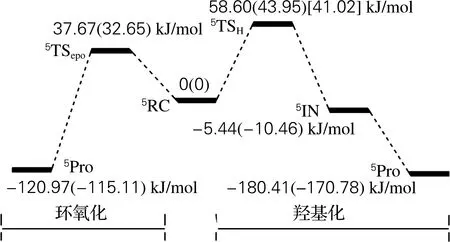
环氧化反应速控步骤是形成C—O键中间体的过程,也就是自由基中间体的形成。图4给出了三种配合物的环氧化的反应能垒,在B2+ZPE计算水平下分别为32.65、16.74和17.80 kJ/mol。优化的几何结构见图5。
在[FeⅣ(O)(TQA)(L)]n+(L=None,AN,OTf-)与丙烯的反应过程中,[FeⅣ(O)(TQA)]2+与丙烯形成的过渡态的C—O键最短,Fe—O键最长。这一现象与五配位配合物的环氧化反应能垒趋势一致。对于这三个体系,∠Fe—O—C接近180°。
[FeⅣ(O)(TQA)(AN)]2+配合物氧化环己烷的过程中,只检测到羟基化产物,而未检测到环氧化产物[10]。这与上文结论似乎是相悖的,这是因为三种配合物羟基化的反应能垒都是高于环氧化的。为了更好地探索整个反应,本文比较了羟基化和环氧化反应的产物能量。如图4所示,所有环氧化反应产物的能量均高于对应的羟基化反应产物的能量。也就是说,虽然环氧化反应的反应能垒低,但是羟基化产物更稳定,环氧化产物往往会分解。
3 结 论
[FeⅣ(O)(TQA)(L)]n+(L=None,AN,OTf-)配合物的基态为五重态,并且具有较高的氧化活性。理论计算结果表明,五配位的配合物[FeⅣ(O)(TQA)]2+三重态和五重态的能量差要远高于六配位的配合物。三种配合物的羟基化和环氧化反应都发生在五重态反应的势能面上。由于电子效应和空间位阻效应的关系,对于氢夺取反应的过渡态,其Fe—O—H的键角在110°~140°,而不是传统的180°左右。对于环氧化反应,其速控步骤的过渡态的Fe—O—C的键角接近180°。对比羟基化反应的反应能垒可以发现,考虑到隧穿效应校正和零点能校正,三种配合物的反应能垒是一致的,说明隧穿效应对六配位配合物的影响较大。通过对比羟基化和环氧化的反应能垒,环氧化的反应能垒要低于羟基化反应,但是反应产物的稳定性也低于羟基化反应的产物,也就是说羟基化产物更稳定,环氧化产物往往会分解。
猜你喜欢
中西医结合肝病杂志(2021年7期)2021-11-30
建材发展导向(2021年7期)2021-07-16
辽宁科技大学学报(2021年1期)2021-05-07
昆明医科大学学报(2021年1期)2021-02-07
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
湖南师范大学学报·自然科学版(2015年5期)2015-10-20
外语学刊(2014年3期)2014-12-03
