
辅酶Q相关性肾病综合征4例报告并文献复习
2022-03-13 13:38张新张冉冉刘娟娟林毅邵乐平张秋业
青岛大学学报(医学版) 2022年1期
关键词:肾病综合征
张新 张冉冉 刘娟娟 林毅 邵乐平 张秋业
[摘要] 目的 了解儿童辅酶Q相关性肾病综合征的临床特征,提高对该疾病的认识。方法 报告4例辅酶Q相关性肾病综合征病儿的临床资料;在PubMed数据库检索相关病例进行文献复习,总结其临床表现、病理特点、基因突变及药物治疗反应等。结果 本文4例病儿均为男性,发病年龄10个月~10岁;1例表现为肾病综合征样蛋白尿,余3例均为糖皮质激素(激素)耐药型肾病综合征。基因检测显示2例为ADCK4基因突变,另2例为COQ2基因突变。3例病儿予辅酶Q10治疗后随访,尿蛋白均减少;另1例未予辅酶Q10治疗,病情进展迅速,最终死亡。PubMed数据库检索出55例ADCK4基因突变及25例COQ2基因突变相关性肾病综合征病儿,分析显示辅酶Q相关性肾病综合征病儿临床多表现为激素耐药型肾病综合征,肾脏病理活检主要为局灶性节段性肾小球硬化,早期口服辅酶Q10治疗可以减轻蛋白尿。结论 辅酶Q相关性肾病综合征发病早,表现为肾病综合征或肾病样蛋白尿,早期应用辅酶Q10治疗有一定的效果;ADCK4基因c.826G>C和c.1035+8C>A复合杂合突变、COQ2基因c.518G>A和c.912+1(IVS5)de1G杂合突变在激素耐药型肾病综合征中均为首次报道。
[关键词] 泛醌;ADCK4基因;COQ2基因;肾病综合征;肾小球硬化症,局灶节段性
[中图分类号] R692
[文献标志码] A
[文章编号] 2096-5532(2022)01-0145-05
doi:10.11712/jms.2096-5532.2022.58.019
肾病综合征(NS)是常见的儿童肾脏疾病,儿童原发性NS年发病率为(2~4)/10万,患病率为16/10万[1]。糖皮质激素(激素)耐药型肾病综合征(SRNS)是儿童终末期肾病的第二常见原因[2]。目前已发现有27种基因与SRNS的发病相关,其中辅酶Q相关的基因突变病儿约占遗传性NS病儿的2%[3-5]。辅酶Q相关性NS是儿科少见/罕见病。文献检索显示,国内文献报道该病12例,中国学者在国外文献报告该病不足80例[3-4, 6-25]。本文对4例确诊的辅酶Q相关性NS病儿临床资料进行回顾性分析,并复习相关文献报道,以提高对该疾病的认识,实现早诊断、早治疗。
1 临床资料
1.1 病例报告
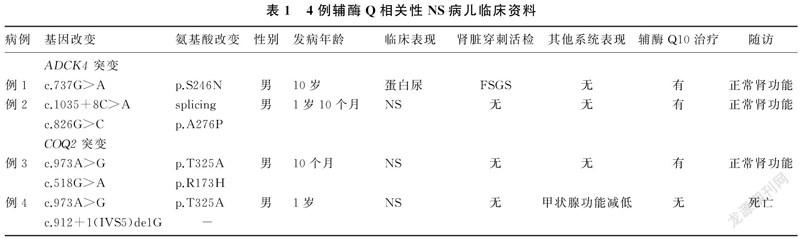
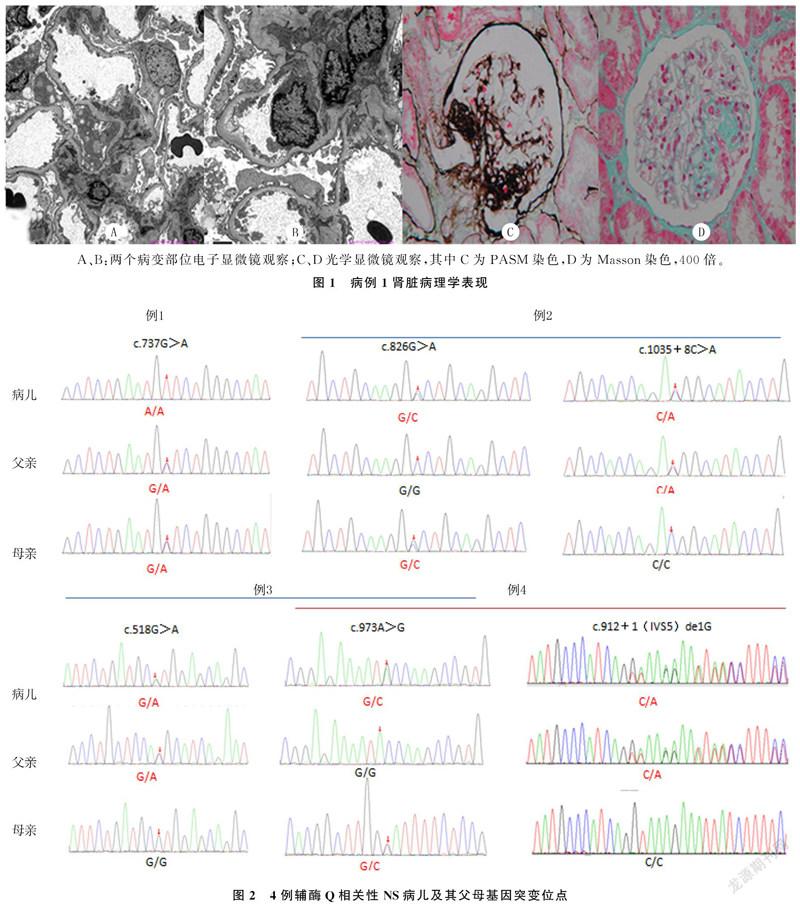
病例1,男,10岁。因“尿检异常1月余”入院。入院前1月因腰痛于当地医院就诊。尿常规示蛋白();血清蛋白35.9 g/L。予口服肾炎康复片治疗6 d,2次随机尿常规检查尿蛋白仍()。入院前10 d泌尿系超声示双肾椎体内钙盐沉积。为进一步诊治转入青岛大学附属医院。既往史无异常。查体:BP 15.9/11.4 kPa,全身无水肿,心、肺、腹部和神经系统无异常。实验室检查:尿蛋白(),24 h尿蛋白定量为61 mg/(kg·d),24 h尿钙定量为0.22 mg/kg;血清清蛋白31.38 g/L,血清胆固醇为4.60 mmol/L;内生肌酐清除率为82.42 mL/(min·1.73 m2);肝炎全套、肾功能、电解质、补体、抗核抗体、尿液细菌培养结果均无异常。肾脏活检病理符合局灶节段性肾小球硬化(FSGS)(图1)。基因检测证实为ADCK4基因c.737G>A纯合突变(来自父母),见表1和图2。根据美国医学遗传学与基因组学学会(ACMG)发布的基因序列变异解读指南、PolyPhen2软件预测并结合临床,判定为致病性基因突变(强致病性证据,强度PS1)。予培哚普利和肾炎康复片治疗近2月,尿蛋白仍为()。基因确诊后加用辅酶Q10(42 mg/(kg·d))治疗。随访观察7个月,随机尿蛋白由()减至(+),监测肾功能无异常。
病例2,男,1岁10个月。因“双眼睑水肿1周”入院。入院前1周出现双眼睑水肿,伴尿量减少,就诊于当地医院,多次随机尿常规示尿蛋白(),转入青岛大学附属医院。既往史无异常。查体:BP 13.8/5.7 kPa,双眼睑水肿;腹膨隆,肝脾未及;心、肺及神经系统查体无异常;双下肢凹陷性水肿。实验室检查:尿蛋白(),隐血(),红细胞73.70 μL;随机尿蛋白/肌酐比值为15;血清清蛋白浓度为19.9 g/L,胆固醇10.58 mmol/L;内生肌酐清除率146 mL/(min·1.73 m2);肝炎全套、抗核抗体、抗中性粒细胞胞浆抗体、抗链球菌溶血素O、红细胞沉降率、补体、肾功能以及电解质等检查均无异常;泌尿系超声示双肾实质回声增强;基因检测证实为ADCK4基因复合杂合突变,分别为c.826G>C、c.1035+8C>A,前者源自母亲,后者源自父亲(表1、图2)。根据ACMG发布的基因序列变异解读指南,用PolyPhen2软件预测并结合临床,判定为致病性突变(中等致病性证据,强度为PM2)。该基因位点突变模式在SRNS中为首次报道。入院后予泼尼松龙2 mg/(kg·d)诱导缓解,同时给予扩容利尿、抗凝和降压等综合治疗,17 d后尿蛋白转阴;但4 d后出现反复,随机尿常规检查示尿蛋白(+)。家属拒绝肾脏穿刺活检。基因确诊后予辅酶Q10(34 mg/(kg·d))治療,同时将激素逐渐减量。随访观察2月余,病儿水肿消退,血清蛋白恢复至正常水平,多次随机尿常规检查显示尿蛋白(±),监测肾功能无受损。
病例3,男,10个月。因“眼睑水肿10余天”就诊于当地医院。入院前10余天出现眼睑水肿伴腹泻,当地医院予蒙脱石散治疗1周,水肿加重。复诊并复查,尿常规示尿蛋白(),血清蛋白6.31 g/L,疑诊NS收入当地医院。入院后复查,晨尿尿常规尿蛋白(),隐血(+),红细胞2.64/μL;血清清蛋白10.8 g/L,肾功能、红细胞沉降率、补体、凝血功能、泌尿系超声均无异常。予泼尼松龙2 mg/(kg·d)诱导治疗,同时予抗感染、抗凝、扩容利尿等对症治疗。治疗10 d水肿加重,蛋白尿仍()。转至青岛大学附属医院。继续给予足量激素诱导缓解治疗3周余,尿蛋白仍波动在()~()。家属拒绝肾脏穿刺活组织病理检查。基因检测证实为COQ2基因复合杂合突变,分别为c.518G>A、c.973A>G,前者来自父亲,后者来自母亲,见表1、图2。根据ACMG发布的基因序列变异解读指南及多种生物信息学蛋白功能预测软件预测并结合临床,判定为致病性突变(致病性证据,c.518G>A强度为PS1+PM2+PP3,c.973A>G为PS1+PP3)。其中COQ2基因c.518G>A杂合突变在SRNS中为首次报道。给予辅酶Q10(1 mg/(kg·d))治疗,同时激素渐减量。随访观察近7个月,病儿水肿消退,随机尿常规检查尿蛋白(+)~(),血清清蛋白恢复达40.3 g/L,未出现肾功能损害。
病例4,男,1岁。因“双眼睑及下肢水肿27 d”入院。入院前27 d出现双眼睑及下肢水肿,就诊于当地医院住院治疗。多次晨尿尿常规示:尿蛋白()~(),隐血(+)~(),红细胞20.0~33.6/HP;血清清蛋白21 g/L,血胆固醇10.79 mmol/L;补体、抗核抗体、ds-DNA抗体无异常;红细胞沉降率、血凝常规、肾功能均正常;泌尿系超声检查示双肾实质回声增强。予泼尼松龙2 mg/(kg·d)诱导缓解治疗13 d,同时给予扩容利尿、抗凝、甲状腺素片等治疗,尿蛋白仍(),水肿加重,尿量进行性减少。转入青岛大学附属医院。转院前已采集静脉血外送行遗传性肾脏病基因检测。既往史无异常。查体:BP 14.6/8.2 kPa,精神差,全身水肿;心率110 min-1,律齐,心音略低钝,无杂音;腹膨隆,肝脾未及,移动性浊音阳性;肺部及神经系统无异常。实验室检查:血清清蛋白浓度为11.50 g/L,总胆固醇8.16 mmol/L,尿素氮10.20 mmol/L,肌酐76.20 μmol/L,血钠127 mmol/L,血钾3.80 mmol/L,血氯109 mmol/L;血凝常规抗凝血酶Ⅲ30.00%,活化部分凝血活酶时间(APTT)64.20 s,APTT比值2.18R,血纤维蛋白原1.58 g/L,余无异常;补体C3为0.30 g/L,补体C4<0.068 g/L;肝炎全套检查无异常。入院后继续予足量激素诱导治疗9 d,同时间断输注清蛋白或血浆,辅助扩容利尿、抗凝、降压、纠正电解质等治疗,水肿加重,伴呼吸急促。因家属强烈要求并签字转上级医院,转诊途中呼吸困难加重而死亡。未予辅酶Q10治疗。基因检测结果证实为COQ2基因c.973 A>G/c.912+1(IVS5)de1G复合杂合突变,其前者来自母亲,后者来源于父亲。见表1和图2。根据ACMG发布的基因序列变异解读指南、多种生物信息学蛋白功能预测软件预测,并结合相关临床资料,判定为致病性基因突变(致病证据,强度为PVS1+PM2)。其中COQ2基因c.912+1(IVS5)de1G杂合突变在SRNS中为首次报道。
1.2 文献复习
1.2.1 ADCK4基因突变相关性肾病病例文献复习 以“ADCK4 gene”或“COQ8B gene”和“nephrotic syndrome”或“proteinuria”为关键词,PubMed数据库检索出文献11篇,含43个家系55例病儿[3,6-15]。55例病儿发病中位年龄为8岁7个月,31例(56.36%)表现为NS,其余24例表现为不同程度的蛋白尿,伴或不伴有血尿、高血压。部分病儿合并其他系统疾病,其中合并甲状腺肿2例,精神发育迟滞3例,外耳畸形1例,癫痫2例,肥厚性心肌病并身材矮小1例,肺动脉高压1例,先天性房间隔缺损1例,乳房良性肿瘤1例。55例中33例行肾脏穿刺活检,27例(81.8%)肾脏病理为FSGS,1例为毛细血管内增生性肾小球肾炎,1例为肾小球球性硬化,2例为系膜增生性肾小球肾炎,1例为弥漫性系膜硬化,1例为球性废弃。55例病儿中31例口服大剂量辅酶Q10(15~30 mg/(kg·d))治疗,随访3~48个月,其中22例反应良好,4例失访,5例病儿未见明显改善,其中3例合并NPHS1基因改变。1例口服小剂量辅酶Q10(4.5 mg/(kg·d))治疗3个月,随访反应良好。55例病儿中有23例进展为终末期肾病(ESRD),其中15例病儿未接受过辅酶Q10治疗。
1.2.2 COQ2基因突变相关性肾病病例复习 以“COQ2 gene”和“nephrotic syndrome”或“proteinuria”为关键词,检索PubMed数据库,检索到相关文献12篇,共報道了19个家系25例病儿[4,15-25]。25例病儿发病年龄从新生儿期至青春期不等,其中15例于新生儿期发病,1例于学龄期(10岁)发病,2例于青春期发病。文献检索出的25例病儿中13例仅累及肾脏系统,其余12例病儿合并进展性脑病、癫痫发作和肌病等神经肌肉系统病变,其中2例同时合并新生儿糖尿病。12例接受了肾脏穿刺活检,11例表现为FSGS(3例为塌陷型FSGS),1例为新月体肾小球肾炎。11例接受了大剂量辅酶Q10(30~60mg/(kg·d))治疗,8例反应良好;3例疗效不明显,快速进展为ESRD甚至死亡。
2 讨 论
SRNS预后差,约50%的该病病儿在15年内发展为ESRD[26-27]。原发性辅酶Q10缺乏相关性肾病是可能用有效药物治疗的遗传性肾病。多种基因突变可导致原发性辅酶Q10缺乏症,其中以ADCK4、COQ2基因常见。ADCK4基因位于染色体19q13.2上,其突变与足突细胞缺失相关,在敲除了斑马鱼、果蝇的ADCK4基因后,可以重现NS的表型[10]。COQ2基因位于染色体4q21-23上,可以清除氧自由基并减少氧化应激反应。在动物模型中,COQ2基因的沉默导致肾脏裂孔隔膜及腔隙通道超微结构异常,导致畸形的线粒体及线粒体自噬现象增多[28]。
本文病例1发病年龄为10岁,仅有大量蛋白尿症状,肾脏活检病理示FSGS。PARK等[9]研究发现,部分ADCK4基因突变相关性肾病病人具有髓质钙沉着现象。本文病例1与其符合,这对辅酶Q相关性NS诊断可能有一定的提示作用。EROGLU等[19]研究显示,COQ2基因突变可引起多系统受累。本文病例3、4均于小婴儿期发病,症状较重,其中病例4同时合并甲状腺功能减低。
本文4例病儿,2例为ADCK4基因突变,2例为COQ2基因突变。通过检索ACMG基因序列变异解读指南及HGMD、OMIM、PubMed等数据库,并采用通用预测软件对突变基因的生物学信息进行分析,结合临床表现,4例均判定为致病性基因突变。已有文献报道,COQ2基因c.973A>G突变及ADCK4基因c.737G>A与NS有关[6-7,23,17]。本文研究中ADCK4基因c.826G>C/c.1035+8C>A复合杂合突变,COQ2基因c.912+1(IVS5)de1G杂合突变和COQ2基因c.518G>A杂合突变在SRNS中均为首次报道,丰富了基因库信息。
輔酶Q相关性肾病常规治疗无效,补充辅酶Q10可防止线粒体形态改变,减少蛋白尿,延缓病情进展[29]。本文病例1未予激素治疗,其余3例对激素抵抗。病例1、2、3基因确诊后予辅酶Q10治疗,其中大剂量(30~60 mg/(kg·d))治疗2例,小剂量(1 mg/(kg·d))治疗1例,随访2~7个月,随机尿常规检查尿蛋白出现不同程度的减少;病例4未早期诊断,病情进展迅速,未予辅酶Q10治疗,最终死亡。复习文献也显示,ADCK4基因及COQ2基因突变所致肾病病儿,应用辅酶Q10均有一定疗效;与ADCK4基因突变病儿相比,COQ2基因突变病儿发病年龄早,易发生死亡结局。提示对于辅酶Q相关性肾病病儿来说,早期诊断及应用辅酶Q10治疗十分关键。但是,目前临床长期服用大剂量辅酶Q10不良反应尚不明确。
综上所述,辅酶Q相关性NS发病早,表现为NS或者肾病样蛋白尿(对激素抵抗),肾脏穿刺活检病理多符合FSGS,早期应用辅酶Q10治疗有一定的效果。
[参考文献]
[1]EDDY A A, SYMONS J M. Nephrotic syndrome in childhood[J]. The Lancet, 2003,362(9384):629-639.
[2]MALAGA-DIEGUEZ L, SUSZTAK K. ADCK4 “reenergizes” nephrotic syndrome[J]. The Journal of Clinical Investigation, 2013,123(12):4996-4999.
[3]LOLIN K, CHIODINI B D, HENNAUT E, et al. Early-onset of ADCK4 glomerulopathy with renal failure: a case report[J]. BMC Medical Genetics, 2017,18(1):28.
[4]STARR M C, CHANG I J, FINN L S, et al. COQ2 nephro-pathy: a treatable cause of nephrotic syndrome in children[J]. Pediatric Nephrology, 2018,33(7):1257-1261.
[5]SADOWSKI C E, LOVRIC S, ASHRAF S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome[J]. Journal of the American Society of Nephrology: JASN, 2015,26(6):1279-1289.
[6]WANG F, ZHANG Y Q, MAO J H, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome[J]. Pediatric Nephrology (Berlin, Germany), 2017,32 (7):1181-1192.
[7]ZHANG H W, WANG F, LIU X Y, et al. Steroid-resistant nephrotic syndrome caused by co-inheritance of mutations at NPHS1 and ADCK4 genes in two Chinese siblings[J]. Intractable & Rare Diseases Research, 2017,6(4):299-303.
[8]YANG J, YANG Y, HU Z X. A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy[J]. Biochemical and Biophysical Research Communications, 2018,506(3):444-449.
[9]PARK E, KANG H G, CHOI Y H, et al. Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations[J]. Pediatric Nephrology (Berlin, Germany), 2017,32(9):1547-1554.
[10]ASHRAF S, GEE H Y, WOERNER S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption[J]. The Journal of Clinical Investigation, 2013,123(12):5179-5189.
[11]FENG C Y, WANG Q, WANG J J, et al. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal di-sease and ADCK4 mutation: case reports and literature review[J]. Medicine, 2017,96(47):e8880.
[12]李国民,孙利,沈茜,等. 线粒体相关肾病2例病例报告及文献复习[J]. 中国循证儿科杂志, 2015,10(6):426-433.
[13]杨钊,顾崇娟,郑旭蕾,等. 一个单纯性蛋白尿家系的致病突变分析[J]. 中华医学遗传学杂志, 2019,36(6):598-601.
[14]宋晓翔,徐虹,沈茜,等. ADCK4相关肾小球病8例分析[J]. 中华肾脏病杂志, 2017(1):22-29.
[15]童桂霞,刘雪梅,张洪霞,等. 以肾病综合征起病的原发性辅酶Q10缺陷1型一例[J]. 中华儿科杂志, 2017,55(3):230.
[16]SCALAIS E, CHAFAI R, VAN COSTER R, et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in Para-hydroxybenzoate-polyprenyl transferase (COQ2)[J]. European Journal of Paediatric Neurology (EJPN): Official Journal of the European Paediatric Neurology Society, 2013,17(6):625-630.
[17]DIOMEDI-CAMASSEI F, DI GIANDOMENICO S, SANTORELLI F M, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement[J]. Journal of the American Society of Nephrology: JASN, 2007,18(10):2773-2780.
[18]QUINZII C, NAINI, SALVIATI L, et al. A mutation in Para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency[J]. American Journal of Human Genetics, 2006,78(2):345-349.
[19]EROGLU F K, OZALTIN F, GN N, et al. Response to early coenzyme Q10 supplementation is not sustained in CoQ10 deficiency caused by CoQ2 mutation[J]. Pediatric Neurology, 2018,88:71-74.
[20]BEZDKA M, TOLBOV , SEEMAN T, et al. Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: a high proportion of causative variants in NUP93[J]. Pediatric Nephrology (Berlin, Germany), 2018,33(8):1347-1363.
[21]GIGANTE M, DIELLA S, SANTANGELO L, et al. Further phenotypic heterogeneity of CoQ10 deficiency associated with steroid resistant nephrotic syndrome and novel COQ2 and COQ6 variants[J]. Clinical Genetics, 2017,92(2):224-226.
[22]WU X, WANG W H, LIU Y, et al. A steroid-resistant nephrotic syndrome in an infant resulting from a consanguineous marriage with COQ2 and ARSB gene mutations: a case report[J]. BMC Medical Genetics, 2019,20(1):165.
[23]徐可,毛曉燕,姚勇,等. COQ2基因变异致婴儿型肾病综合征一例临床分析并文献复习[J]. 中华儿科杂志, 2018,56(9):662-666.
[24]LPEZ L C, SCHUELKE M, QUINZII C M, et al. Leigh Syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations[J]. The American Journal of Human Genetics, 2006,79(6):1125-1129.
[25]BEZDKA M, DLUHOLUCKY M, CINEK O, et al. Successful maintenance of partial remission in a child with COQ2 nephropathy by coenzyme Q10 treatment[J]. Nephrology (Carlton, Vic), 2020,25(2):187-188.
[26]TRAUTMANN A, BODRIA M, OZALTIN F, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort[J]. Clinical Journal of the American Society of Nephrology: CJASN, 2015,10(4):592-600.
[27]LOVRIC S, ASHRAF S, TAN W Z, et al. Genetic testing in steroid-resistant nephrotic syndrome: when and how[J]? Nephrology Dialysis Transplantation, 2016,31(11):1802-1813.
[28]ZHU J Y, FU Y L, RICHMAN A, et al. A personalized mo-del of COQ2 nephropathy rescued by the wild-type COQ2 allele or dietary coenzyme Q10 supplementation[J]. Journal of the American Society of Nephrology, 2017,28(9):2607-2617.
[29]PERSSON M F, FRANZN S, CATRINA S B, et al. Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes[J]. Diabetologia, 2012,55(5):1535-1543.
(本文编辑 黄建乡)
3481500338241
猜你喜欢
中国现代医生(2016年31期)2017-03-02
中外医疗(2016年31期)2017-02-28
现代仪器与医疗(2016年6期)2017-01-12
糖尿病新世界(2016年16期)2016-12-09
上海医药(2016年19期)2016-11-09
中国实用医药(2016年16期)2016-07-26
中国实用医药(2016年15期)2016-05-24
现代养生·下半月(2015年9期)2015-09-28
医学信息(2015年6期)2015-03-17
