
基于ceRNA调控网络的乳腺癌预后相关生物标志物筛选
2022-05-19 03:54刘俊彤王培培叶明全黎青青
右江民族医学院学报 2022年2期
刘俊彤,王培培,叶明全,黎青青
(1. 皖南医学院医学信息学院,安徽 芜湖 241002;2. 皖南医学院健康大数据挖掘与应用研究中心,安徽 芜湖 241002)
乳腺癌是全球范围内女性常见的癌症之一,约占全球新发癌症病例的11.7%,也是造成女性患癌死亡的主要原因之一[1]。近年来,乳腺癌的发病率呈逐年上升的趋势[2]。而早期的乳腺癌并没有典型的临床表现,一旦确诊往往已是中晚期[3],因此,通过寻找高特异性的乳腺癌生物标志物,对改善患者的预后及生存率具有十分重要的意义。
随着RNA高通测序技术的发展,转录组数据呈爆发式增长,信使RNA(messageRNA,mRNA)一直是主要研究重点,是蛋白质的合成模板[4]。非编码蛋白质的RNA主要包括微小RNA(microRNA,miRNA)和长链非编码RNA(long non-coding RNA,lncRNA),二者可以通过与蛋白质、DNA、RNA相互作用,参与基因表达的调控[5],已有研究表明microRNA和lncRNA可以通过多种方式影响癌基因的表达[6-8],但是,目前肿瘤预后相关生物标志物的挖掘主要基于mRNA、miRNA、lncRNA中的一种分子标签,而忽略了mRNA-miRNA-lncRNA之间的相互调控作用以及它们对癌症的共同作用。
本研究利用对癌症公共数据库(TCGA)的挖掘,分别筛选乳腺癌预后相关的mRNA、miRNA和lncRNA分子标签,综合考虑每种标签的可靠性和标签之间的关联,构建基于mRNA-miRNA-lncRNA的竞争性内源RNA(competing endogenous RNA,ceRNA)网络,同时探究调控网络中的mRNA与免疫系统的相关性,提出的核心ceRNA调控网络(3个mRNA、1个lncRNA、2个miRNA)为乳腺癌的预后研究提供了新的研究方向,同时本研究的模型也可用于其他肿瘤预后相关标志物的筛选参考。
1 材料与方法
1.1 TCGA数据下载和预处理 从TCGA数据库(https://portal.gdc.cancer.gov/)获得乳腺癌的转录组测序序列(RNA-seq)、miRNA测序序列(miRNA-seq)以及临床信息,包括1 078例BRCA样本和104个正常对照。乳腺癌的项目ID为TCGA-BRCA。据检索得到的数据按以下条件筛选:①有完整且可供分析的生存数据(包括生存状态、总生存时间);②每一个样本的mRNA数据、lncRNA数据以及miRNA数据完整。最后对符合标准患者的测序数据和生存信息下载和整合,以供进一步分析。
1.2 差异基因筛选及可视化 利用R包“Deseq2”对基因数据的差异表达进行分析,筛选出 |Log2 Fold change| >1和P值< 0.05的基因。使用R包“pheatmap”分别绘制差异表达mRNA、lncRNA、miRNA的聚类热图,使用R包“EnhancedVolcano”分别绘制差异表达mRNA、lncRNA、miRNA的火山图。
1.3 ceRNA网络构建及可视化 Cytoscape是一个基于Java的可视化平台,可以整合分子交互网络与基因表达数据和其它分子的状态信息。利用miRcode数据库对特征miRNA调控的lncRNA进行分析预测,利用miRTarBase、starbase和miRDB等数据库对miRNA调控的靶基因进行分析预测,筛选符合ceRNA机制的基因,筛选标准如下:利用Pearson相关系数计算mRNA与lncRNA的相关性,筛选出表达正相关的pairs(P值<0.01);根据ceRNA网络理论,lncRNA和mRNA竞争性结合miRNA,二者之间必须共享大量的miRNA,因此,通过超几何检验来判断基因数据间是否存在显著水平的多miRNA共享,设定超几何检验的P值<0.01满足筛选条件。将得到的网络关系导入Cytoscape中绘制ceRNA网络调控关系图。
1.4 ceRNA网络核心RNA预后分析 构建ceRNA网络后,为了进一步明确网络中的差异mRNA、lncRNA和miRNA与乳腺癌预后的关系,我们采用R包“Survival”进行单因素COX回归分析,作出Kaplan-Meier生存曲线,并设定P值<0.05具有统计学意义。
2 结果
2.1 乳腺癌预后相关的mRNA分子标签 从TCGA下载乳腺癌患者的RNA测序序列(RNA-seq)以及临床信息,挑选其中RNA-seq数据中“group”列为protein_coding的数据作为编码蛋白的mRNA表达数据,使用“Deseq2”进行差异表达分析,筛选log2FC绝对值>1和Pvalue<0.01认为是差异表达基因。与癌旁正常组织比较,共筛选出2 767个编码蛋白差异表达mRNA,其中,上调1 125个,下调1 642个。使用R包“pheatmap”和“EnhancedVolcano”对已筛选差异表达基因的热图和火山图分别进行绘制,结果见图1,图1A为热图,红色表示高表达,蓝色表示低表达,横轴代表样本,样本聚类显示在横轴上方,纵轴代表mRNA。图1B为火山图,Log2值>1的基因以蓝色标记;Log2值>2的基因以红色标记。为进一步探究差异基因可能涉及的生物学功能和通路,我们进行了通路富集分析。富集分析结果如图2所示,乳腺癌组织中上调mRNA富集通路的前5名是细胞周期、细胞分裂、含CENPA核小体在着丝粒上的沉积、细胞周期过程调控、癌症中的视网膜母细胞瘤基因;乳腺癌组织中下调mRNA富集通路的前5名是血管生成、肌肉结构发育、细胞成分运动的正向调节、循环系统过程、阿米巴型细胞迁移。利用单因素COX比例风险回归模型分析与生存期显著相关的mRNA,Log-rank检验结果的P值<0.05的共306个基因,其中,与生存期正相关103个,负相关203个。表1显示与生存期相关性最高的前20个mRNA。
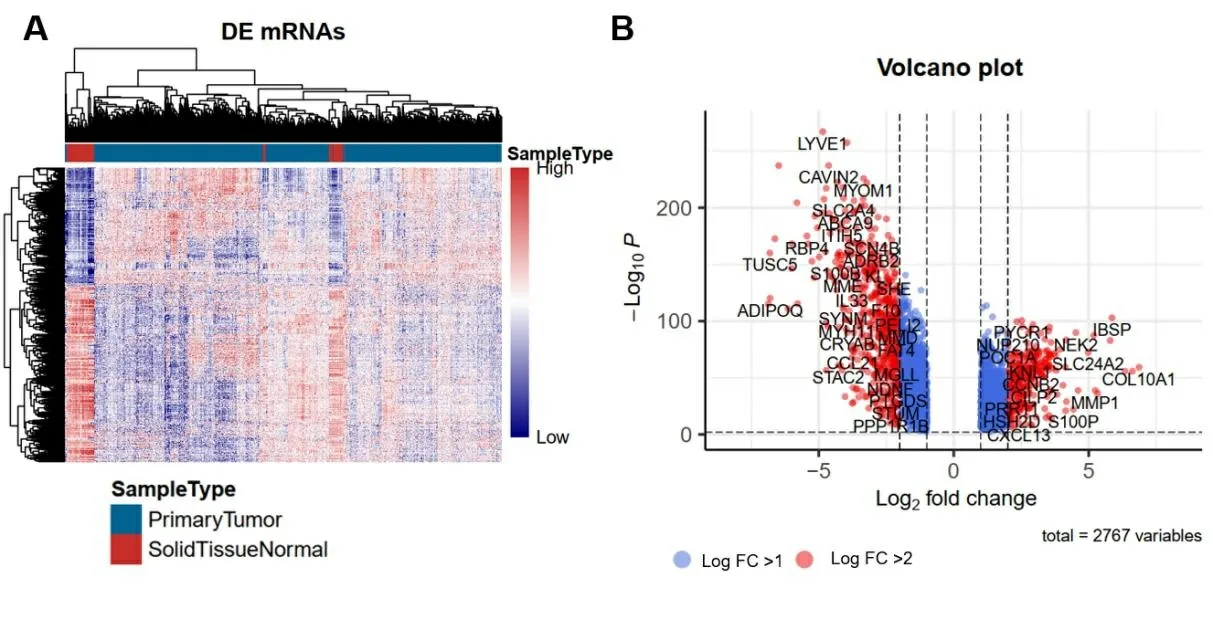
图1 差异表达的mRNA可视化
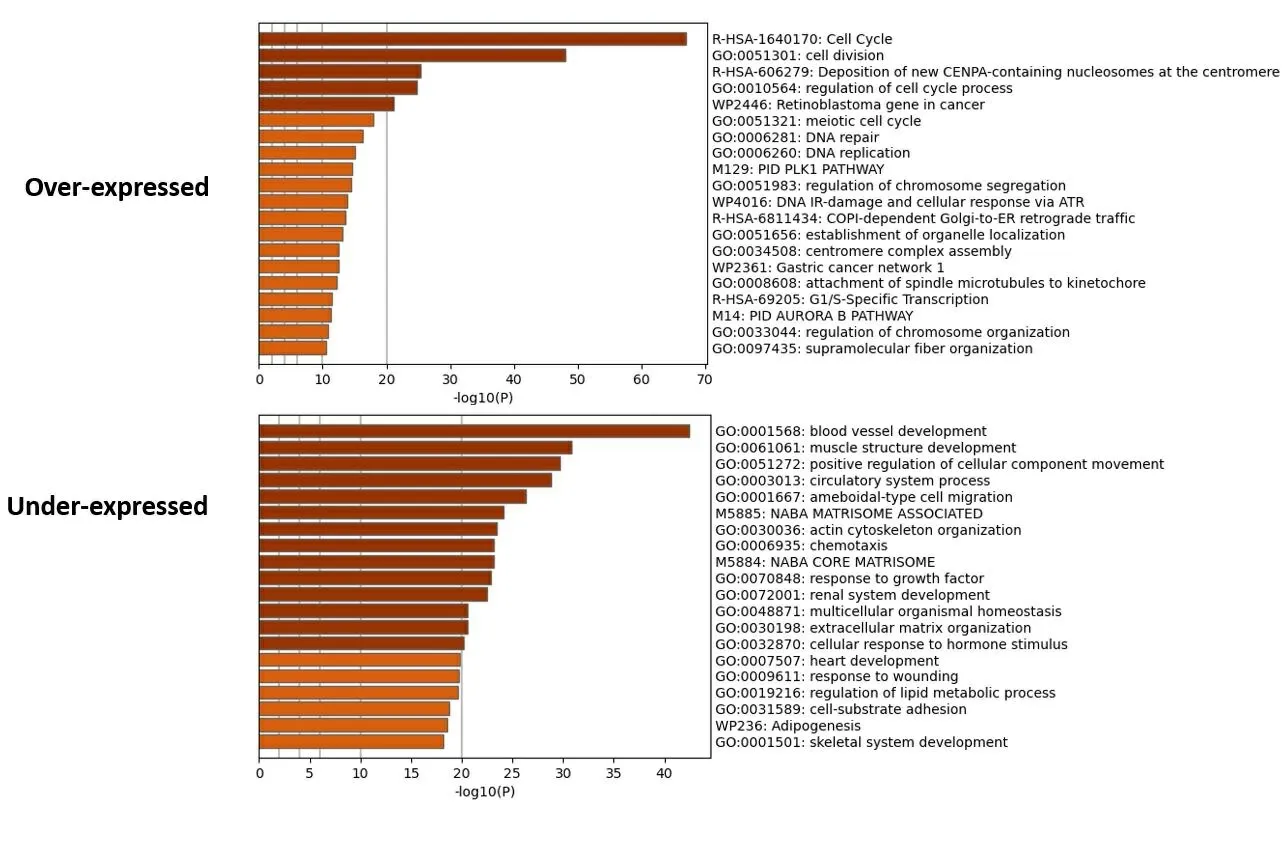
图2 乳腺癌差异表达上调和下调mRNA通路富集结果
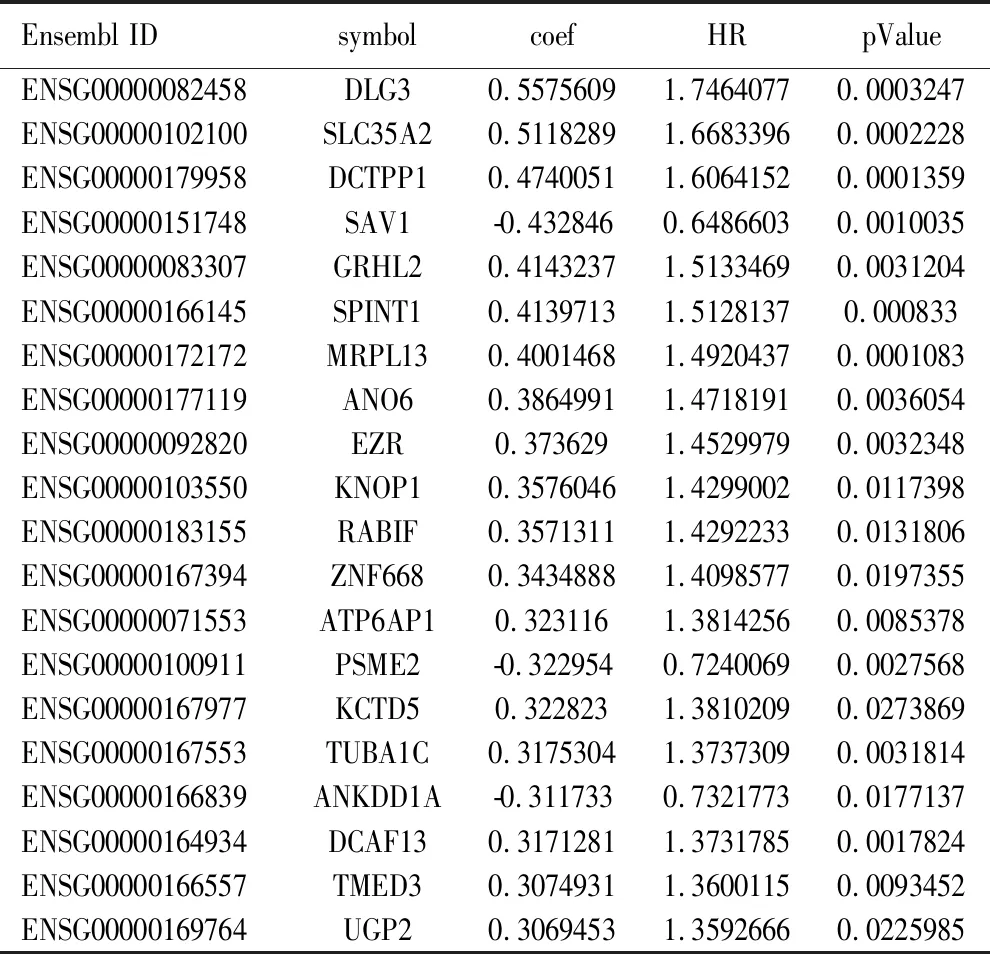
表1 与乳腺癌生存期相关性最高的前20个mRNA
2.2 乳腺癌预后相关的LncRNA分子标签 与癌旁正常组织比较,共筛选出167个差异表达的lncRNA,其中上调61个,下调106个。差异表达lncRNA的热图和火山图见图3。利用单因素COX比例风险回归模型分析与生存期显著相关的lncRNA,Log-rank检验结果的P值<0.05的共18个基因,其中,与生存期正相关2个,负相关16个。表2显示与生存期显著相关性的前18个lncRNA。
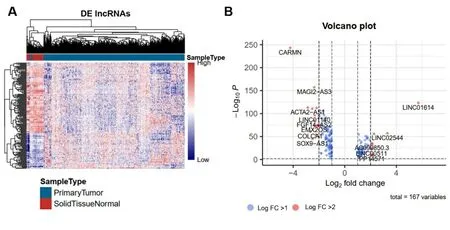
图3 差异表达的lncRNA可视化

表2 与乳腺癌生存期具有显著相关性的18个lncRNA
2.3 乳腺癌预后相关的miRNA分子标签 与癌旁正常组织比较,共筛选出158个差异表达的miRNA,其中上调71个,下调87个。差异表达miRNA的热图和火山图见图4。利用单因素COX比例风险回归模型分析与生存期显著相关的mRNA,Log-rank检验结果的P值<0.05的共14个基因,其中,与生存期正相关4个,负相关10个。表3显示与生存期显著相关性的前14个miRNA。

图4 差异表达的miRNA可视化
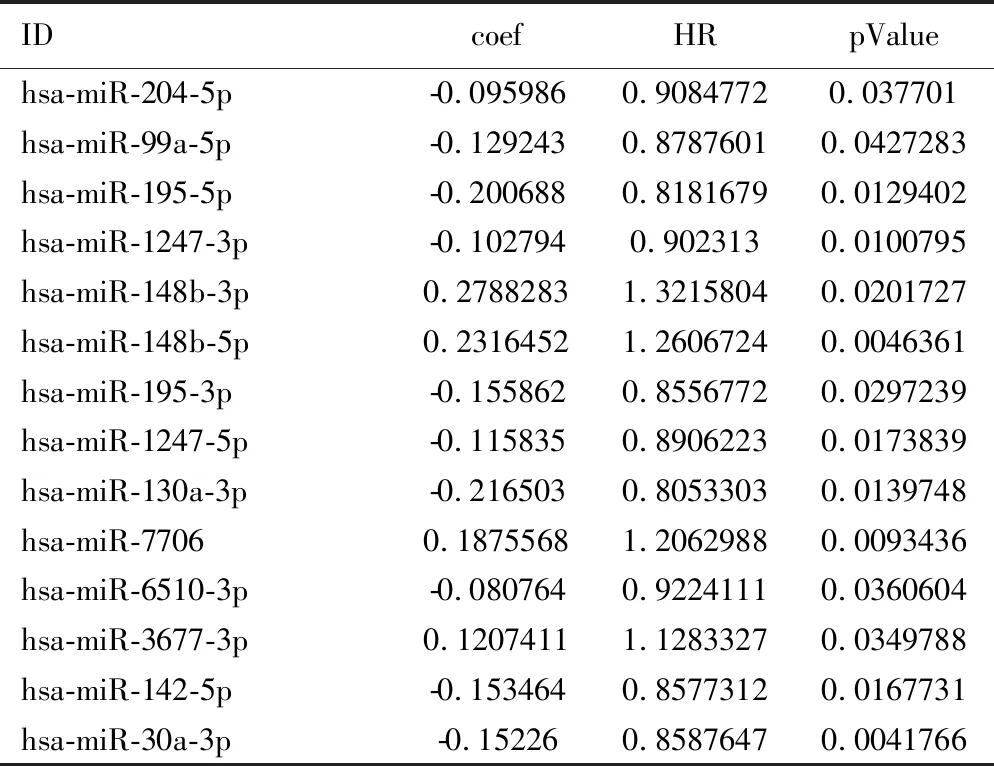
表3 与乳腺癌生存期具有显著相关性的14个miRNA
2.4 乳腺癌ceRNA调控网络的构建 为了更好地理解与生存期密切相关的差异RNAs在乳腺癌中的作用,本研究通过ceRNA机制进一步确认这些RNAs的相互关系和生物学功能。借助miRcode数据库对lncRNA和miRNA之间的关系进行分析预测,miRNA和mRNA之间的作用关系通过starBase数据库完成预测,整理结果后对网络进行可视化。将构建的mRNA-miRNA-lncRNA关系结果导入Cytoscape软件,绘制调控网络图,如图5所示。其中图5A为利用差异且生存相关mRNA为中心构建的ceRNA,图5B为利用差异且生存相关的mRNA和差异miRNA、lncRNA为中心构建的ceRNA,图5C为利用差异且生存相关的mRNA、miRNA和lncRNA为中心构建的ceRNA。随着筛选条件的提高,核心子网中的基因越来越少,最终为3个mRNA、1个lncRNA和2个miRNA。
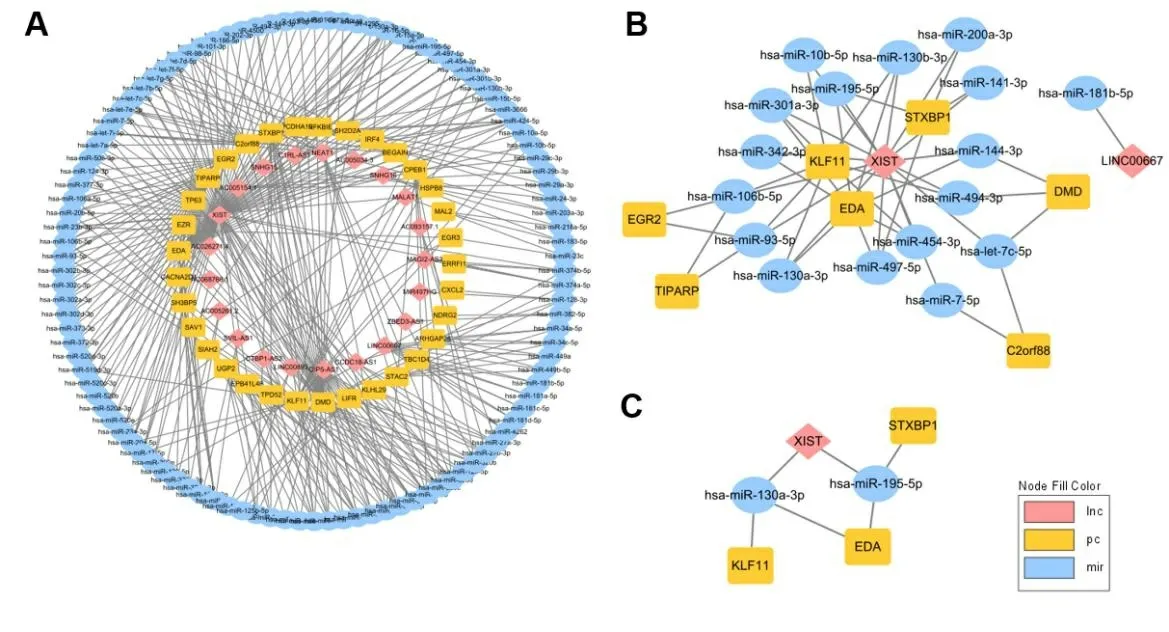
图5 乳腺癌的ceRNA网络
2.5 乳腺癌ceRNA网络核心RNA功能分析 我们使用R包“survival”对ceRNA核心网络3个mRNA(KLF11、EDA、STXBP1)、1个lncRNA(XIST)和2个miRNA(hsa-miR-130a-3p、hsa-miR-195-5p)分别进行生存分析,生存曲线结果见图6。结果表明,hsa-miR-130a-3p、hsa-miR-195-5p两个miRNA高表达,总生存期长;mRNA和lncRNA高表达,总生存期短。乳腺癌根据病理亚型分为Luminal A型、Luminal B型、Her2阳性和三阴性乳腺癌,因同时具有明确分子分型、基因测序数据、临床数据的样本量较少,部分分型甚至出现无样本情况,我们将预后最差的三阴性乳腺癌作为一组,其他三种分型合并为一组,对核心ceRNA子网络中的6个RNA进一步做生存分析,生存曲线结果见图7。结果能表明在不同乳腺癌分子分型中,已筛选核心子网RNA对生存周期的影响具有一致性。因为miRNA和lncRNA在体内主要是调控mRNA作用,接下来我们对核心ceRNA网络中的mRNA进行了通路分析,如表4。结果表明,KLF11和EDA与免疫系统功能相关,而STXBP1主要与突触和神经递质功能相关。利用GEPIA工具,我们对3个mRNA(KLF11、EDA、STXBP1)组成的分子标签与免疫系统功能相关性进行探究见图8。结果表明KLF11、EDA、STXBP1组成的分子标签与效应T细胞和驻留记忆T细胞存在显著的正相关性。效应T细胞分泌的穿孔素可以溶解肿瘤细胞膜或者被感染的细胞膜,进而导致靶细胞形成孔洞并最终解体。而记忆T细胞保留了一些对抗原的免疫记忆,当抗原再次入侵时,可迅速增值分化产生大量的效应T细胞和记忆T细胞,再次破坏靶细胞并释放出其中的抗原。
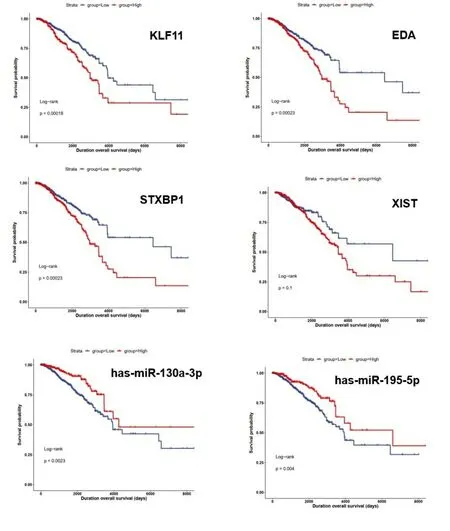
图6 乳腺癌ceRNA核心子网RNA的生存分析

图7 三阴性乳腺癌/非三阴性乳腺癌ceRNA核心子网RNA的生存分析

表4 乳腺癌ceRNA核心子网mRNA的通路分析
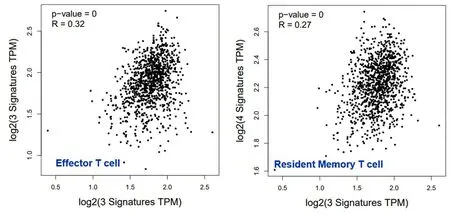
图8 乳腺癌ceRNA核心子网mRNA标签与免疫细胞的相关性分析
3 讨论
随着乳腺癌发病率的逐年增加,寻找具有高特异性的乳腺癌生物标志物,对改善乳腺癌患者的预后及生存率具有重要的意义[9-10]。近年来,基于公共癌症数据库,挖掘乳腺癌预后相关分子标签的研究逐渐成为国内外研究热点[11-13]。
在该研究中,通过对TCGA的挖掘,使用R包“Deseq2”进行差异表达分析,筛选出差异表达基因(2767个mRNA、167个lncRNA、158个miRNA),利用单因素COX比例风险回归模型对差异表达基因进行生存分析,筛选出差异表达且生存相关基因(306个mRNA、18个lncRNA、14个miRNA),借助miRcode与starBase数据库,设定超几何检验的P值<0.01满足筛选条件,将得到的网络关系导入Cytoscape中绘制ceRNA网络调控关系图,最终筛选出3个mRNA(KLF11、EDA、STXBP1)、1个lncRNA(XIST)和2个miRNA(hsa-miR-130a-3p、hsa-miR-195-5p)参与构建ceRNA网络,使用R包“survival”对ceRNA核心网的6个RNA进行生存分析,结果表明mRNA和lncRNA高表达、miRNA低表达,会促使乳腺癌进一步增值和侵袭,相互调控关系见图9,对核心ceRNA网络中mRNA的进一步通路分析和免疫分析也验证了筛选出的生物标志物是有意义的。

图9 乳腺癌ceRNA核心子网RNA的在乳腺癌增值、侵袭的作用
本研究获得了一个有意义的乳腺癌ceRNA核心子网,其中心分子相互调节并在乳腺癌中发挥重要作用,有望成为潜在的治疗靶点,同时本研究的策略为其它肿瘤预后相关标志物的筛选提供了新的思路。进一步拟通过生物学实验探索和验证这些RNA在乳腺癌各个分子分型发展的机制。
猜你喜欢
理财周刊(2022年4期)2022-04-30
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
学苑创造·A版(2020年12期)2020-01-07
大学教育(2019年5期)2019-04-23
中国新通信(2019年21期)2019-03-30
价值工程(2018年35期)2018-01-25
物流科技(2017年10期)2017-11-22
中国现代医生(2016年31期)2017-03-02
中国现代医生(2016年32期)2017-03-02
