
不同铈盐体系(硝酸盐、硫酸盐、氯化盐)中CTAB与Ce3+的相互作用机理
2022-06-24 07:58孙德贇胡艳宏刘鹏唐茂胡泽柳召刚吴锦绣
化工进展 2022年6期
孙德贇,胡艳宏,刘鹏,唐茂,胡泽,柳召刚,吴锦绣
(1 内蒙古科技大学材料与冶金学院,内蒙古 包头 014000;2 内蒙古自治区稀土湿法冶金与轻稀土应用重点实验室,内蒙古 包头 014000;3 轻稀土资源绿色提取与高效利用教育部重点实验室,内蒙古 包头 014000)
包头白云鄂博稀土矿资源世界第一,轻稀土占98%以上,而其中的50%为铈资源。二氧化铈具有变价性(+3、+4两种价态)、光谱吸收性能、耐磨性、高折射率等,是一种用途极为广泛的稀土类氧化物。在紫外吸收方面,可添加至化妆品中,起保护皮肤的作用;在光催化领域,不同粒径、比表面积的二氧化铈能作为优良的催化剂,被广泛应用于光催化领域;在玻璃抛光和添加领域,不同粒径尺寸的二氧化铈可作为抛光粉,用于玻璃表面抛光,不同形貌的二氧化铈可作为玻璃添加剂,增加其自身的透光性;在工业废气催化领域,二氧化铈可作为载体,在负载不同活性的物质(如Cu、Co、Ni、Mn 等)后,能提高催化剂的活化性能,增强对甲苯等有害气体的催化。但很多领域都要求其粒子具有特殊的物性,以提高材料的性能。所以,特殊物性稀土化合物的产业化制备技术的开发就显得十分重要。在这些制备的方法中,由于模板法具有简单快捷、成本低以及可控性强等优点,正在逐渐被越来越多的专家和学者青睐。
随着科技进步和交叉学科发展,计算机理论计算合成新材料的方法越来越受科学家们的重视,从头算量子化学方法能够从原子或分子尺度研究材料的形核机理和生长方式,解决实验中不能解释的机理,半经验的分子动力学方法计算时,采用以牛顿运动方程为基础的经验势函数方法,描绘介观体系下晶体中每个晶面的生长过程,从而能够动态地了解晶体在特定条件下的生长状态。迄今为止,无论采用何种方法制备二氧化铈,较多采用硝酸铈作为铈源,也并未发现前人对于为什么较多采用硝酸铈作为铈源作出系统的解释。本文采用密度泛函理论(DFT)和第一性原理研究的方法,模拟模板法制备二氧化铈时,选用工业中常用的萃取工艺完成后可直接使用的3 种不同铈盐体系(硝酸盐、硫酸盐、氯化盐)探究模板剂十六烷基三甲基溴化铵(CTAB)在水环境中与Ce的相互作用机理,通过分析不同铈盐体系下各原子间形成化学键的COHP、ICOHP等,从原子角度解释了CTAB在3 种不同铈盐环境下对Ce的控制作用,从而简要说明采用硝酸铈作为铈源制备二氧化铈时在形核阶段的优势。
1 模型与方法
1.1 计算方法
本文采用Vienna Ab Initio 模拟软件包,使用广义梯度近似(GGA)方法执行DFT 计算,并使用涉及化学键分析的LOBSTER 软件包研究3种不同体系下模板剂CTAB 与Ce的相互作用以及化学键的影响。由于Ce 在碳酸铈晶体中是以+3 价的离子态存在的,使得4f 轨道中只有一个电子,且4f 轨道的能量通常明显低于5d 轨道,并伴随着轨道径向分布极度收缩,表现出类似半核的特性,因此不参与成键。出于简洁的需要,采用PBEsol泛函分别计算了考虑到f轨道电子的PBEsol+U赝势以及把f轨道的电子当作内层电子计算的简化赝势的态密度(DOS) 和各元素的分波态密度(PDOS),如图1 所示。在不考虑峰强条件下,发现这两种方式计算出的电子结构除了Ce 以外,C、H、O 的总态密度和分波态密度所产生峰的位置没有太大误差;DFT+U 与简化赝势相比,发现在Ce的6s 和5p 轨道中,自旋向上的α 轨道和自旋向下的β轨道能量并不相等,在费米能级附近自旋向上的α 轨道有f 和d 轨道的DOS 而β 轨道上则没有,并且展现出了f 轨道100%自旋极化,这是因为Ce并非是纯的+3价,其4f轨道中的电子有部分离域,所以在PDOS上会出现混合的4f和5d的特征,并且f轨道会对6s和5p轨道产生相应的影响使得其α轨道和β 轨道的能量不相等,这样虽然混合的f-d 轨道处于费米能级附近,但是因为f轨道只有一个电子占据,能量较低,所以不会与晶胞中的其他元素有相互作用,因此在总DOS 图中就会体现出差别不大的态密度。针对上述情况考虑到计算效率,计算方法上选择采用把Ce的f轨道电子当作是内层电子的简化赝势;泛函选用PBEsol泛函;其他的相关计算参数为:平面波截断能ENCUT 为350eV,布里渊区K 点以网格大小为4×3×2 的Gamma 点划分,赝势文件Ce选择为+3价的简化赝势,其他元素如H、O、S、Cl、N、C、Br 的赝势选择+1、-2、-2、-1、+5、+4、-1 价的PAW 赝势。直至两个迭代步骤之间的能量差稳定低于10eV,受力稳定小于-0.05eV/Å(1Å=0.1nm)。考虑整个系统的自旋极化,简化赝势中Ce外层电子为6s5p,不存在孤电子,因此该系统可以由单个分子轨道Ψ表示,可将其视为闭壳系统。整个计算过程都是在0K 下用原子间静力进行优化,优化完成后的结构是相对稳定的基态结构,与实验值相似度较高。

图1 PBEsol泛函计算的DOS和PDOS图
对于化学键的分析,本文使用LOBSTER 软件包计算COHP 以及ICOHP 的方法。COHP 可以从能带结构计算结果中获得成键信息,可以确定材料中成对原子间的成键、非键和反键的相互作用。所谓COHP即哈密顿矩阵乘以相应的态密度矩阵,计算公式如式(1)所示。

式中,f代表占据数;ε代表能带能量;代表原子;代表原子轨道;代表能带(分子轨道);H代表哈密顿矩阵元;N()代表DOS(态密度)矩阵元。
成键贡献使体系的能量降低,COHP 为负值;反键贡献使体系能量升高,COHP为正值;而非键贡献则用COHP 零值表示。实际应用中常用COHP的正值、负值和零来表示成键、反键和非键相互作用。对整个占据轨道COHP 的积分通常表示为ICOHP,可以对成对原子间的成键强度进行定量分析,其定义式(2)。

单独分析ICOHP 的绝对数值没有任何意义,只有进行比较分析其相对数值才能体现出ICOHP的意义,ICOHP 的值越小,成键的稳定性越强,反之则弱。
1.2 建模


图2 球棒模型表示的slab模型中的离子、原子、分子
2 结果与讨论
2.1 硝酸体系内部成键作用分析


图3 优化前后硝酸体系slab模型

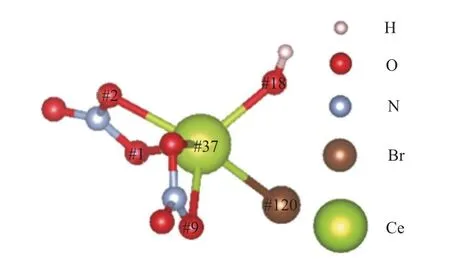
图4 #37_Ce优化完成后Br-、NO、H2O与Ce3+成键结构图

图5 #37_Ce与周围成键原子的COHP和ICOHP图

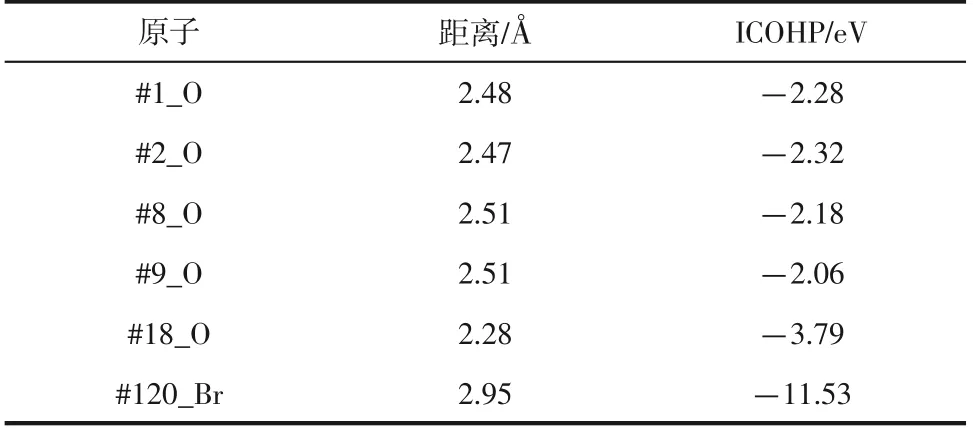
表1 优化完成后#37_Ce与周围原子的键长及ICOHP值
2.2 硫酸体系内部成键作用分析


图6 优化前后硫酸体系slab模型


图7 硫酸体系优化完成后Br-、SO、H2O与Ce3+成键结构图

图8 硫酸体系#27_Ce与周围成键原子的COHP和ICOHP图
2.3 氯化体系内部成键作用分析
在构建的氯化体系中,Ce、CTAB、Cl和HO随机分布在整个slab模型中[图9(a)],优化完成后,如图9(b),Ce周围聚集体系中的Br、HO 以及Cl,其中CTAB 中的Br、HO 中的O以及Cl会围绕着中心Ce发生相互作用,并伴有成键趋势。随着各原子向着受力最小的位置移动,致使整个体系中的原子会向着Ce位置聚集,形成季铵盐络合物。

图9 优化前后氯化体系slab模型
利用Vienna Ab Initio 模拟软件包对氯化体系slab 模型优化完成后#8_Ce 与周围原子的成键情况如 图10 所 示,#8_Ce 分 别 与HO 中 的#13_O、#14_O、#20_O、#25_O、#28_O 形成Ce—O 键,与体系中的#6_Cl形成Ce—Cl 键,与CTAB 中的Br形成Ce—Br 键,采用涉及化学键分析的LOBSTER软件包对各原子间的成键稳定性进行分析。#8_Ce原子与HO 中#13_O、#14_O、#20_O、#25_O、#28_O 形 成Ce—O 键 的-COHP 和ICOHP 的 值 如图11(a)~(e)所示,从COHP的图中可以看出Ce—O键在费米能级以下位置出现能量为负的-COHP值,表明Ce—O键在临近费米能级以下的位置有不稳定成分存在,观察其ICOHP的曲线发现所有的Ce—O键的ICOHP 的曲线几乎都在坐标0 点的右侧,且ICOHP 的值都小于-2,表明Ce与水分子形成的Ce—O键可以比较稳定的存在于体系中,但与硝酸体系、硫酸体系中Ce 与HO 形成的Ce—O 键相比稳定性要弱。#8_Ce 与体系中#6_Cl 的-COHP 及ICOHP 的值如图11(f)所示,从图中可以看出其-COHP 的曲线在-5eV 和-20eV 左右的位置出现了-COHP 为负的峰值,但ICOHP 的曲线都存在于坐标0 点的右侧且ICOHP 的值为-2.48eV,说明Ce—Cl 键虽然在两个位置存在不稳定的成分,但是对成键稳定的影响非常小,Ce—Cl 键可以比较稳定地存在于体系中。体系中#8_Ce 与CTAB 中#131_Br 所 形 成Ce—Br 键 的-COHP 及ICOHP 的 值如图11(g)所示,从整体上来看,费米能级以上-COHP的峰值都处于坐标0点的左侧,费米能级以下-COHP 的峰几乎都处在坐标0 点的右侧,但是在-16eV、-18eV以及-33.5eV左右出现了-COHP的值为负的峰,但峰值很小,对Ce—Br键的成键稳定性的影响可以忽略,ICOHP曲线都位于坐标0点的右侧,且ICOHP 的值达到了-5.63eV,说明在氯化体系中Ce—Br键比Ce—O键、Ce—Cl键更稳定。

图10 氯化体系优化完成后H2O、Br-、Cl-与Ce3+成键结构图

图11 氯化体系#27_Ce与周围成键原子的COHP和ICOHP图
2.4 不同体系下成键比较
不同体系下Ce 与HO 的成键情况如表2 所示。从表中可以看出,硝酸体系和硫酸体系中Ce—O键的ICOHP值小于氯化体系中Ce—O键的ICOHP值,结合上文中COHP值可以得出不同体系的水环境中硝酸体系、硫酸体系比氯化体系中Ce 与HO 形成Ce—O 键稳定性强,但从Ce 与HO 的成键数目可以看出,氯化体系与水的络合能力要远强于硝酸体系和硫酸体系,更易形成络合物。Ce与CTAB的成键情况如表3所示,Ce只与CTAB中的Br有成键作用,在不同体系中Ce—Br 键的键长差距较小,但其ICOHP 的值具有较大差距。图12 为不同铈盐水环境下的Ce与CTAB 分子间的径向分布函数图,可以看出3个不同体系中Ce—Br之间径向分布函数峰值出现的位置极其相近,大约在3Å,但硝酸体系的径向分布函数峰值最高。根据ICOHP 的值以及径向分布函数的峰值大小,可以得出硝酸体系水环境中Ce—Br 键的稳定性最强,表明在硝酸体系中CTAB对Ce的控制能力最强。

图12 不同铈盐水环境下的Ce3+与CTAB分子间的径向分布函数图

表2 不同体系中Ce与H2O的成键情况

表3 不同体系中Ce与CTAB的成键情况
3 结论
本文采用Vienna Ab Initio 模拟软件包以及涉及化学键分析的LOBSTER 软件包等软件,以计算机模拟的手段对添加CTAB 模板剂的3 种不同铈盐体系(硝酸盐、硫酸盐、氯化盐)形核前各原子间成键作用进行了分析。结果证明,3个不同体系中的Ce与HO 形成Ce—O 键的ICOHP 值相差不大,且在-COHP 曲线中出现不稳定成分的位置也极为相似,从而说明在不同体系水溶液环境中Ce与HO形成Ce—O 键的稳定性差距较小,但氯化体系与HO的络合能力要远强于硝酸体系和硫酸体系,更易形成络合物;Ce与CTAB中Br形成Ce—Br键的稳定性差距明显,比较3个体系中Ce—Br键的ICOHP值,硝酸体系(-11.53eV)<硫酸体系(-6.86eV)<氯化体系(-5.63eV),说明硝酸体系中Ce—Br键的稳定性最强,氯化体系中Ce—Br 键的稳定性最弱,可以进一步说明CTAB 在硝酸体系下对Ce具有较强的控制作用,进而证明模板剂CTAB在硝酸体系下对制备二氧化铈晶体形核阶段具有较好的调控能力。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中国食品(2021年18期)2021-09-28
中学化学(2019年2期)2019-07-08
理科考试研究·高中(2017年7期)2017-11-04
中学生理科应试(2017年2期)2017-04-01
中学生数理化·高三版(2016年9期)2016-05-14
数理化学习·高一二版(2009年5期)2009-07-31
数理化学习·高一二版(2009年5期)2009-07-31
