
探讨价层电子对互斥理论的电子对数计算简易方法与应用
2022-07-30 03:23张卫马荔谢少艾汪羽翎陈虹锦
大学化学 2022年6期
张卫,马荔,谢少艾,汪羽翎,陈虹锦
上海交通大学化学化工学院,上海 200240
价层电子对互斥理论(简称VSEPR法)最初是由西奇威克(N. Sidgwick)和鲍威尔(H. M. Powell)在1940年提出的,通过实验他们发现某些特定的分子结构可以简单地与价层电子的数量相关联,60年代初吉莱斯皮(R. J. Gillespie)和尼霍尔姆(R. S. Nyholm)发展并完善了这一理论。VSEPR法非常简单,基于路易斯的共用电子对理论,不需要原子轨道的概念,在解释、判断和预测分子结构的准确性方面与杂化轨道理论比毫不逊色,成为定性处理分子结构问题的有用工具[1]。尽管VSEPR法只适用于讨论AXn型(A为中心原子、X为配位原子、n为配位原子的数目)孤立的共价型分子,且只能对分子的构型作定性的描述,不能说明分子形成原理及键的相对稳定性,但VSEPR法为理解分子几何模型提供了有用的基础,由于其简单易懂,VSEPR法模型作为一种教学工具被纳入教科书中,在中高等化学教育中具有不可替代的地位[2]。
1 VSEPR法的基本规则
1.1 VSEPR法判断分子空间构型的基本流程
VSEPR法运用了电子密度区域的概念将其简化,电子密度区域运用一个球体代表价层电子对,该球形区域表示在该区域价层电子对出现的概率大而在区域之外价层电子对几乎不出现[3],即假设中心原子A的内层电子是球形分布的,不影响价层电子对的排布方式,其周围配置的原子或原子团的几何构型,主要取决于中心原子价电子层中电子对(包括成键电子对和未成键的孤电子对)的互相排斥作用,分子的空间构型总是尽可能采取电子对对称排布且相互排斥力最小的那种结构,若配位原子的数目n与电子对数相等时,其分子构型与电子对构型一致。而当n小于电子对对数时,则分子的空间构型与电子对的几何排布构型不同,按斥力最小的方式确定其空间构型,VSEPR法判断分子空间构型的基本流程见图1。

图1 VSEPR法判断分子结构的基本流程
当分子或基团中存在孤电子对时,需要考虑孤电子对、成键电子对之间相互排斥力的大小,确定排斥力最小的稳定结构,并估计这种结构对理想几何构型的偏离程度。电子对间排斥力大小次序为:孤对电子-孤对电子>孤对电子-键对电子>键对电子-键对电子>键对电子-孤电子,主要考虑电子对间呈90°角度时排斥力。
价层电子对数是应用VSEPR法首先需要确定的问题,高校的《无机化学》《普通化学》等教材在计算中心原子的价电子对数时常采用两大类方法,一类是[4-6]采用写下分子或离子的Lewis结构式并识别出中心原子,通过化合价确定孤对电子数目,然后通过配位原子个数和孤对电子数确定价层电子对数,由于需要准确画出Lewis结构式,在复杂体系中应用比较困难;另一类是[7,8]做了规定:作为中心原子时提供价电子数为其价层电子数,而作为配位原子时,卤族原子和氢原子提供1个价电子,氧族原子不提供价电子,当作提供价电子数为0,这类方法非常简便,通过确定中心原子和配位原子后,应用配位原子提供价电子的约定,能非常方便地计算出价层电子总数,但因未解释作为配位原子时卤族原子提供1个价电子、而氧族原子不提供价电子的原因,学生在应用过程中会有疑问,同时也没有明确说明N和C作为配位原子时提供的价电子数。另外,有学者[9]研究不区分中心原子和配位原子利用公式直接计算价层电子对数的方法,对于多原子分子体系不利于后续确定分子的空间结构及判断化学键的组成;还有学者[10]提出一种符合化合价原则的定构价层电子对概念,根据逻辑学按照电子对的不同属性对中心原子所有的电子对进行系统分类以判断分子构型,这种方法基于掌握化合物的中心原子和配位原子间的化学键组成,实际应用较为困难。
下面根据路易斯的共用电子对理论,即八隅律,非氢原子周围8电子为稳定结构,氢原子周围2电子时为稳定结构,通过价层电子对数计算方法,讨论不同主族元素作为配位原子时相当于提供的电子数,进一步明确作为配位原子时卤族原子提供1个价电子、而氧族原子不提供价电子的原因,另一方面也明确N和C作为配位原子时提供的价电子数,从而完善中心原子和配位原子提供价电子数的计算规则不同、更简便地计算价层电子对数的方法。
1.2 配位原子相当于提供价电子数的推导
设a为中心原子的价电子数,x为配位原子的价电子数,n为配位原子的个数,P为中心原子的价层电子对数,对于AXn型共价分子而言,其价层电子对数P包括未成对电子对和成对电子对,当配位原子不为氢时,未成对电子对数可用总价电子数之和(中心原子和配位原子提供的)减去配位原子满8电子稳定结构的电子数,二者差除以2计算,成对电子对数为配位原子的个数,即:
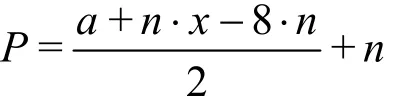
化简得:
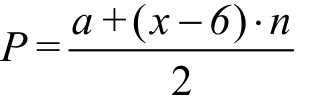
从化简结果可以看出,价电子总数包括两个部分,其一为中心原子的价电子数(即a),另一部分为n个配位原子提供的,即每个配位原子相当于提供x- 6个价电子来进行计算,令x- 6 =t,当VIIA元素作为配位原子时t值为1,当VIA元素作为配位原子(如O)时t值为0,相当于不提供价电子,而当VA元素作为配位原子(如N)时t值为-1,将p区非金属元素作为中心原子提供的价电子数a及作为配位原子时相当于提供的价电子数t汇总表1中。

表1 p区非金属元素价层电子数的计算规则
容易推导出,若配位原子为H时,AHn的价电子对数P则为:
即得H作为配位原子时提供的价电子数为1。
利用表1的计算规则就能非常简便地计算出价层电子对数,在应用价层电子对互斥理论分子的空间构型等时非常便捷。
2 VSEPR法的应用讨论
2.1 直接应用示例
对于配位原子同时包含H和非H原子的共价型化合物,采用表1中心原子和配位原子提供价电子规则计算价层电子对数更为简便,比如三氯甲烷CHCl3,中心原子C提供4个价电子,配位原子H、Cl都相当于提供1个价电子,价层电子对数P= 4,分子呈正四面体构型;而直接用八隅律方法,各原子提供的价层总电子数为26,按非H原子的8电子、H原子的2电子稳定结构,价层电子对数,或者采用26除以8的商为3余数为2,其中商值代表非H配位原子的电子对数,将余数再除以2得到H配位原子的电子对数(或孤电子对数),此例中价层电子对数为3 + 1 = 4,通过对比可以看出将利用表1计算规则确定价层电子对数非常简便。
需要注意的是,VSEPR法只能简单判断出分子的空间构型。中心原子A与配位原子X之间可能是单键,也可能是多重键。由于在判别非过渡元素共价化合物分子(或离子基团)的空间构型时,VSEPR法与杂化轨道理论的结果基本一致,表明两种方法间具有一定的联系,杂化轨道形成的σ键决定了分子的骨架,未参与杂化的p轨道从侧面重迭形成π键,不影响分子的基本几何形状。杂化轨道数等于价层电子对数,二者在确定分子的空间构型上具有一致性,二者都是以电子配对理论为基础的,只是考虑问题的角度不同而已[11]。这样就可以通过VSEPR法计算价层电子对数,从而推断出中心原子的杂化类型,通过具体的杂化过程,推断中心原子和配位原子间化学键的组成,进一步确定键角的大小。
2.2 判断键角的大小
当AXn型共价分子中存在孤对电子、中心原子与配位原子间存在多重键、电负性差异等情况时,分子的键角会相对于对称的价电子对几何构型有小幅度的改变,如对于H2O、OF2、SF2三个三原子组成的共价化合物,计算其价层电子对数都为4对,存在两个孤对电子,分子的空间构型都为V型,比较其键角的大小需要根据中心原子A或配位原子X电负性的不同,先比较H2O、OF2,二者中心原子都是O,配位原子分别为H、F,F的电负性大,成键电子对相对远离O原子,孤对电子受到成键电子对的斥力减小,键角小些,即∠HOH大于∠FOF;类似地,OF2、SF2的配位原子相同,中心原子不同,因O原子电负性大,成键电子对更靠近O,孤对电子受到成键电子对的排斥力增大,键角大些,即∠FOF大于∠FSF,故H2O、OF2、SF2三个V型分子的键角依次减小。需要注意的是,在讨论电负性对分子键角大小的影响时,各分子中心原子的价层电子对数要相同,即电子对的几何构型一致,这样键角受电负性影响变化的基础范围是一致的。
另外,在利用电负性差异判断键角大小时,若成键电对间包含有双键或大π键,还要同时考虑双键或大π键的差异。比如判断SO2和臭氧O3分子的键角大小,VSEPR法计算出价层电子对数P为3,即杂化轨道数目亦为3,推断其杂化类型为sp2,中心原子S的价层电子排布为3s23p4,杂化过程如图2所示。
在S、O间形成σ键的同时,中心原子S未参与杂化的3p轨道中2个电子与在每个O原子2p轨道中尚有一个单电子会肩并肩形成大π键,即三个原子共享4个电子的π键,表示为π43。再判断O3分子空间构型,可将其中的一个O原子作为中心原子,其他2个O看做配位原子,即写成OO2型,利用表1计算规则,可方便地计算出价层电子对数为3,中心原子O为sp2杂化,分子呈V型结构,与SO2分子类似,也存在三个O原子共享4个电子的π键,即π43,与SO2分子比较,配位原子相同,仅考虑电负性差异判断,理论上臭氧O3分子因其中心原子O的电负性大于S,其键角应大于SO2分子的键角,但实验测得SO2分子的键角为119.5°、O3分子的键角为116.8°,O3分子的键角要小些,这是由于两分子中的成键电子对都并非单键,而是存在共享电子的π43键,因此需要考虑多重键的键能大小对键角的影响,查得SO2、O2分子键能[12]分别为552 ± 8、498.36 ± 0.17 kJ·mol-1,由于O3分子较O2更活泼,其键能低于O2分子,因此可推断出SO2分子中成键电子对间的排斥作用更大,键角较O3分子的大,同时由于两分子中中心原子和配位原子间都不是单键结构,孤对电子的排斥作用不显著,分子的键角较3个价电子对形成平面正三角形的键角结构(120°)下降幅度都较小。
2.3 判断链状结构有机小分子的杂化类型
由于表1的价电子计算规则区分了中心原子和配位原子通过价电子数的差异,对于链状结构有机小分子骨架上原子的杂化类型的判断也非常简便,如乙烯CH2=CH2,作为中心原子的C提供价电子数为4,配位原子的H、C相当于提供的价电子数分别为1、-2,则价层电子总数= 4 + 1 × 4 + (-2) = 6,价层电子对数P= 3,其杂化类型为sp2。类似地,可以计算出乙炔CH≡CH的中心原子C的价层电子总数= 4 + 1 × 2 + (-2) = 4,价层电子对数P= 2,其杂化类型为sp。对于骨架上超过3个原子的链状结构,在计算中心原子价层电子总数时,对于不是作为直接相连的配位原子,按其与配位原子相连的结构,单键、双键、三键分别作为价电子数1、2、3计入价层电子总数,以丙酸CH3CH2COOH为例加以说明,链的骨架上有3个C原子,以羧基中的C为中心原子研究时,按表1规则,中心原子C提供价电子数为4,两个氧提供价电子为0,H提供价电子为1,―CH2―作为配位原子则提供价电子数= -2 + 2 = 0,CH3―以单键与配位原子C相连,提供的价电子数以1计,即可得到价层电子总数= 6,价层电子对数P= 3,该C原子的杂化类型为sp2,将丙酸CH3CH2COOH中各C原子的杂化类型判断汇总于表2中。
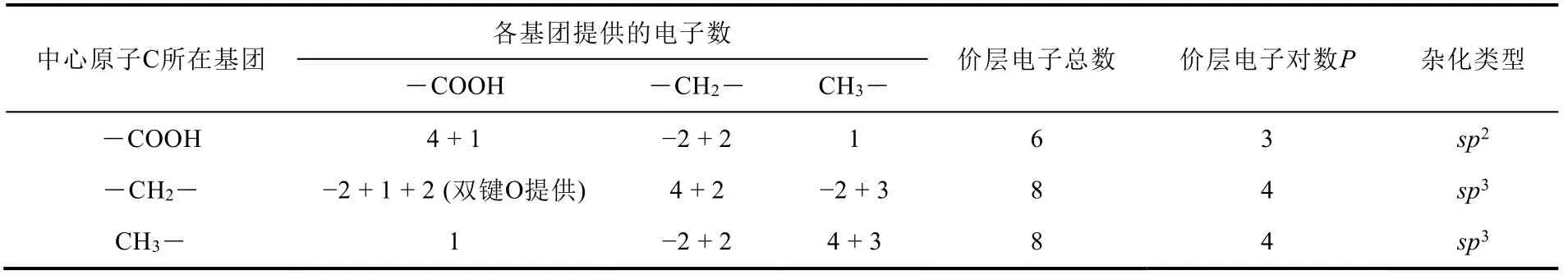
表2 VSEPR法判断丙酸CH3CH2COOH中各C原子杂化类型
2.4 长周期p区非金属元素的应用
短周期p区非金属元素共价化合物的空间构型及键角大小利用VSEPR法和杂化轨道理论进行预测和判断,与实验事实具有较高的一致性,但长周期的p区非金属元素共价化合物利用该方法,常有理论预测与实际不相符合的情况,比如第五周期的Sb(VA)、Te(VIA)形成的配阴离子[SbCl6]3-、[TeBr6]2-,按照表1方法计算出两个基团的价层电子对数都为7,用VSEPR法判断二者都是变形的八面体结构,但实验测得其为正八面体结构,相当于价层电子对数都为6。这是由于第五周期p区元素的价电子层中5s2电子钻穿作用强,不易于参与成键,相当于在计算价层电子对数时需要考虑减去这一对电子的作用,这样修正后的VSEPR法判断的分子构型就与实际相符了。
3 结语
价层电子对数的确定是应用VSEPR法预测、判断、解释AXn型共价分子空间构型的基础,通过数学公式推导确定了p区非金属元素作为配位原子时相当于提供的价电子数,一方面解释了作为配位原子时卤族原子提供1个价电子、而氧族原子不提供价电子的原因,另一方面也明确N和C作为配位原子时相当于提供的价电子数分别为-1和-2,从而完善将中心原子和配位原子按不同计算规则、更简便地计算价层电子对数的方法。同时采用简便价层电子对计算规则讨论了链状结构有机小分子的杂化类型的判断方法,长周期p区非金属元素的最外层s电子的钻穿作用对价层电子总数的影响,也探讨了VSEPR法与杂化轨道理论联用确定中心原子与配位原子间化学键的组成,以及利用电负性差异比较共价型分子键角大小时需要注意多重键的影响等,这些都是对VSEPR法应用的有益补充。
VSEPR法在无机化学、大学化学的教学中具有不可替代的地位,理解并掌握价层电子对数的确定,对定性研究分子空间结构有着重要的应用价值。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
新世纪智能(数学备考)(2021年9期)2021-11-24
新世纪智能(数学备考)(2020年9期)2021-01-04
语数外学习·高中版上旬(2020年8期)2020-09-10
当代陕西(2019年6期)2019-04-17
中学生数理化·高一版(2018年10期)2018-11-08
分析化学(2018年2期)2018-03-02
读写算·教研版(2016年8期)2016-05-07
外语学刊(2014年3期)2014-12-03
