
FDA医疗组合产品上市前申请路径
2022-08-03 03:38林源
中国医疗器械杂志 2022年4期
【作 者】林源
Potrero医疗股份有限公司,海沃德市,加利福尼亚州,美国,94545
0 引言
医疗组合产品为众多医学问题提供了独特的解决方案,但相对复杂的产品组成也带来了监管方面的挑战。美国食品和药物管理局(Food and Drug Administration,FDA)从20世纪70年代开始监管组合产品的申报[1],在过去50多年中积累了丰富的经验,也进行了多次申报程序的改进,目前形成了相对清晰和完整的监管方案。2022年1月,FDA发布了最新的关于组合产品上市前路径指导原则文件[2]。文件给出当前FDA对于组合产品上市前审查原则的思考,提供了包括组合产品的定义,申请人和FDA、FDA内部机构之间关于组合产品监管的沟通和协调,FDA对于组合产品上市前的审查,以及如何确定适当的组合产品上市前申请路径等宏观的综合性信息。而申请路径的确认通常是组合产品上市前准备的第一步也是最重要的一步。下面结合此指导原则,针对组合产品上市前申请路径进行深入分析。
1 组合产品的定义和涵盖范围
《联邦食品药品化妆品法案》[3]和《美国联邦法规》[4]明确给出了医疗组合产品的定义,即两种及以上药品、医疗器械和/或生物制品组成的医疗产品。组合产品中包含的药品、器械和生物制品被称为组合产品的“组成部分”。
组合产品的涵盖范围:
(1)“单一实体”组合产品:由两种及以上受监管成分组成,以物理、化学或其他方式组合或混合的“单一实体”组合产品,包括作为单一实体生产的医疗器械/药品、医疗器械/生物制品、生物制品/药品或医疗器械/药品/生物制品。例如预装注射器或药品洗脱支架。
(2)“共同包装”组合产品:两种及以上独立产品“共同包装”的组合产品,例如包括止血胶布和抗菌药品的手术器械包或急救箱。
(3)与已批准产品“联合使用”的组合产品:某种单独包装的待审批的医疗器械,药品或生物制品,根据其研究计划或使用说明,仅可与已经获得批准的、特殊指定的医疗器械、药品或生物制品一起使用。这两种产品均是达到预期用途和治疗效果的必要组成部分。在新产品批准后,已批准的产品需要根据用途、适应证和效果更新产品说明,以反映预期用途、剂型、强度、给药途径或剂量的显著变化。例如光活化药品,可与单独指定的激光源一起治疗皮肤疾病。
(4)其他“联合使用”的组合产品:某单独包装的待审批医疗器械、药物或生物制品,仅可与另一种特殊指定的待审批的医疗器械、药品或生物制品一起使用。这两种产品均是达到预期用途和治疗效果的必需组成部分。
2 组合产品的监管机构
组合产品的审批由三个审评中心参与,包括医疗器械和放射卫生中心(Center for Devices and Radiological Health,CDRH)、药品评估和研究中心(Center of Drug Evaluation and Research,CDER)和生物制品审评与研究中心(Center for Biologics Evaluation and Research,CBER)。组合产品的监管要求源自适用于医疗器械、药品和生物制品的法律法规要求,当它们是组合产品的组成部分时,将保持它们原有的监管属性[5]。组合产品的专门监管规定通常旨在解决适用于医疗器械、药品和生物制品组成部分的法律和监管要求之间的重复和区别。
1990年以前,FDA没有明确的组合产品监管方法。当两个或多个中心对组合产品负有审查责任并需考虑采用多套法规时,管辖权分配和审查流程等方面都存在不确定性。1990年,美国国会通过《安全医疗器械法案》[6],要求组合产品上市前审查和监管管辖权由指定的一个审评中心主导,并根据组合产品的首要作用方式(primary mode of action,PMOA)来确定管辖权的分配。首要作用方式是指该组合产品中的某个单一组成部分(医疗器械、药品或生物制品)提供了的最重要的治疗作用,即确定该组合产品在所有疗效中起到最大贡献的作用方式[3,7]。例如,如果医疗器械/生物制品组合产品的PMOA可归因于生物制品,则负责此类生物制品上市前审查的中心将对组合产品的监管具有主要管辖权。
为进一步提高组合产品审批的效率和质量,FDA依据《医疗器械使用者费用和现代化法案》[8]于2002年建立了组合产品办公室(Office of Combination Products,OCP)。目前OCP的主要职责包括[9]:
(1)针对组合产品问题,作为FDA工作人员和行业的主要联系人。
(2)制定指南、法规和标准操作程序,以明确组合产品法规。
(3)将产品分类为医疗器械、药品、生物制品或组合产品,并指定相应的评审中心进行上市前审查和上市后安全监督。
(4)通过监督和协调多个审评中心,确保组合产品及时和有效的审查。
(5)监督上市后安全性,以保护患者免受于目前市场上的组合产品相关的潜在健康风险。
(6)协助及时解决有关组合产品上市前审查的争议。
(7)更新关于组合产品的协议、指导文件或实践。
(8)就办公室的行动和效果向国会提交年度报告。
(9)向FDA工作人员和受监管行业提供关于组合产品监管的培训。
3 组合产品的上市前申请路径的确认流程
3.1 单一上市前申请
《联邦食品药品化妆品法案》第503(g)(1)(B)和503(g)(6)节规定,FDA“应在适当的情况下接受任何组合产品的单一上市前申请”[3]。当然,除非FDA确定单一申请是必要的,申请人可以选择为组合产品的不同组成部分提交单独的申请。FDA目前的想法是单一申请通常适用于组合产品,以简化与FDA的监管互动并避免多个申请可能引发的不必要重复。但是,跨标签组合产品的组成部分通常允许单独申请,在使用这种方法时,申请人应与两个相关审评中心进行协调(并可能请求OCP协助),以促进有效、及时地考虑可能与每个组成部分以及它们的组合使用相关的问题。单一申请将简化组合产品上市申请以及和FDA直接的沟通,并避免分开申请各组成部分时可能引起的不必要重复。单一申请应包括每个组成部分的资料,以便对每个组成成分进行安全性和有效性评估。
3.2 主审中心的确认
根据组合产品的首要作用方式,OCP将确定该组合产品的主审中心。组合产品的申请逐年上升,表1是对2015-2020年度各审评中心受理组合产品的汇总[10],可以看到医疗器械和放射卫生中心受理的申请数量占比最大,综合6年数据,约为总体组合产品的65%。
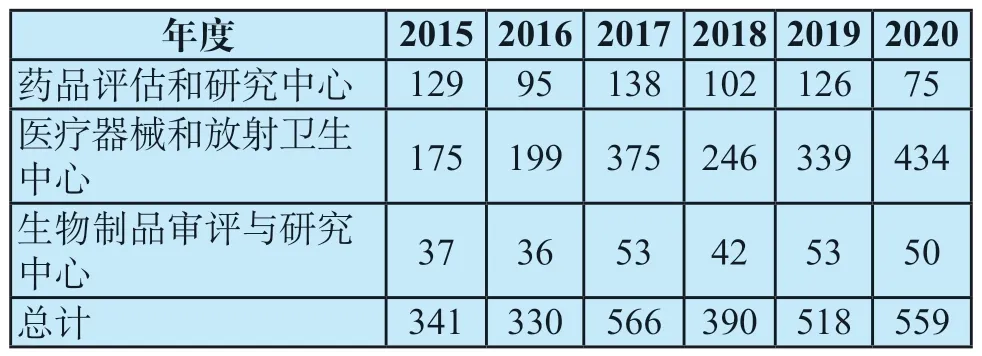
表1 2015—2020年各审评中心受理的组合产品申请数量[n]Tab.1 Number of combination product applications accepted by each center from 2015 to 2020
3.2.1 产品属性界定申请
产品属性界定申请(Request for Designation,RFD)是FDA帮助申请人在提交上市申请之前认定产品属性、分类和申请路径的途径,同样也适用于当申请人对组合产品的管辖权不明确时,要求FDA评估和指定对产品具有主要管辖权的主审中心。申请人可以通过产品属性界定申请获得正式且具有约束力的管辖权决定。根据FDA的《行业和员工指南:如何撰写属性界定申请》[11],产品属性界定申请需以书面申请形式递交,并包括以下信息,以帮助FDA评估组合产品的首要作用方式:
(1)发起人的身份,包括公司名称和地址、设立登记号码、公司联系人和电话号码。
(2)产品说明,包括产品的分类、名称、组成部分、各组成部分目前的获批情况、申请人与监管机构之间有关将此产品的任何讨论或协议、研发进展、制造过程(包括所有组成部分的来源)、拟定用途、已知作用方式、申请人对该产品最重要的治疗作用的单一作用方式的建议以及该决定的依据、使用时间和期限、药品或生物制品的剂量和给药途径以及相关产品及其监管状况等信息。
(3)申请人关于哪个审评机构应具有主要管辖权的建议。
OCP必须在收到RFD之内的5个工作日之内确认申请是否完整,并向申请人发送确认提交日期的通知,或通知申请人RFD未完成提交,并确定完成RFD提交所需的信息。OCP需在提交日期的60天内做出最终答复。如果在该时间段内未收到回复,则申请人在RFD中所建议的产品分类和主审中心即被采纳。若申请人对结果有异议,需在收到确认信的15天内要求OCP重新考虑。
表2总结了2015—2020年度RFD接收和审批的情况[10,12-16],可以看出材料不全的申请比例非常高,6年平均达到80%,而获得最终确认结果的只有13.7%,比例相对较低。因此,申请人在准备申请时需要深入了解RFD的申请要求,如果有任何疑问可以在申请前联系OCP进行沟通。

表2 2015—2020年度RFD接收和审批的情况[n]Tab.2 RFD receipt and approval status from 2015 to 2020
表3总结了2017—2020年度RFD审批的时间[10,14-16],所有获得结论的RFD全部在60天时限之内完成,且各年度完成时间基本一致。因此用60天作为预估RFD从收到确认提交日期的通知到获得结论是比较准确的。
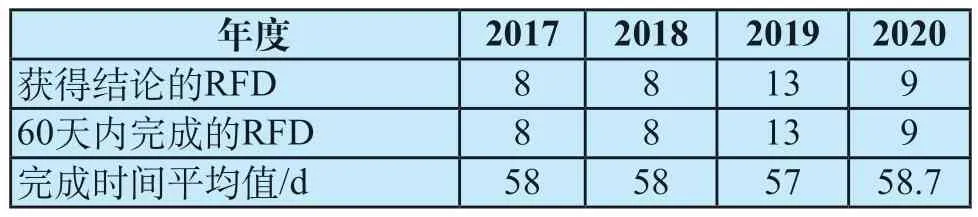
表3 2017—2020年度RFD审批时间Tab.3 RFD approval time from 2017 to 2020
3.2.2 产品属性界定预申请
自2002年12月24日成立以来,OCP一直为申请人在产品开发的不同阶段提供帮助。申请人经常寻求OCP反馈,以了解他们的医疗产品是否将作为医疗器械、药品、生物制品或组合产品进行监管,以及哪个监管中心(CDRH、CDER或CBER)将对其进行监管。申请人可以通过两种方式从OCP接收此类反馈,一种是提交RFD,另一种灵活的选择是向OCP提交咨询请求而得到初步的不具有约束力的管辖权评估。随着时间的推移,申请人越来越倾向于使用这种非正式的方式获取反馈。同时,FDA也希望提高该非正式流程的透明度和一致性,于是在2016年开始正式实施产品属性界定预申请(Pre-RFD),并于2018年发布了“产品属性界定预申请指南”[17]。该指南为申请人提供了清晰的结构化流程和明确的申请建议,还提供了FDA工作人员审查Pre-RFD的流程,申请人从OCP接收反馈的一般时间表,以及安排与Pre-RFD相关的会议。如果申请人对Pre-RFD的申请有任何疑问,FDA鼓励申请人在提交Pre-RFD之前与OCP联系和沟通。虽然对产品分类和管辖权分配的Pre-RFD没有规定的回复时间,但是,OCP会尝试在与RFD相同的时间范围内(即60天内)回复。由于Pre-RFD的灵活性,申请人对其的利用率高于RFD,图1所示为2017—2020年度RFD与Pre-RFD提交数量的对比。

图1 2017—2020年度RFD与Pre-RFD提交数量的对比Fig.1 Comparison of RFD and Pre-RFD submissions from 2017 to 2020
Pre-RFD可在任何时候进行提交,尤其在研发早期,产品分类或主审中心不清晰或存在争议时,FDA的指导信息将有助于申请人提前做出合理的决策。当然,如果产品有明确规定的分类和管辖权分配,则不需要Pre-RFD。OCP在其网站上会不定时公布一些常见的组合产品示例,包括药物洗脱支架等,为申请人提供实用的参考信息[18]。
4 组合产品的上市前申请路径
4.1 医疗器械主导型组合产品
医疗器械主导型组合产品通过上市批准(premarket approval,PMA)、新型医疗器械分类申请(De Novo)以及上市前通知(510(k))来进行上市前申请。
4.1.1 上市批准
Ⅲ类医疗器械一般都必须在上市前获得上市批准(PMA),医疗器械主导型组合产品也不例外。PMA的材料应包含可以充分证明组合产品整体安全性和有效性的数据。在批准或拒绝 PMA之前,相关的FDA咨询委员会可以在公开会议上审查PMA,并向FDA提供是否应批准该申请的建议。
4.1.2 新型医疗器械分类申请(De Novo)
FDA没有分类过的医疗器械自动划归Ⅲ类医疗器械。如申请人有异议,认为产品应该分类为I或Ⅱ类,可提交新型医疗器械分类申请(De Novo)。如果申请人能证明其产品符合《联邦食品药品化妆品法案》第513(a)中的标准[19],FDA会批准其分类要求,并发出产品分类的书面证明。如果产品不符合I类或Ⅱ类医疗器械的规定,则新分类申请被拒绝,该产品仍被归类为Ⅲ类,必须经过PMA批准流程。
4.1.3 上市前通知(510(k))
上市前通知(510(k))的审核标准是待审批产品与已上市的对比产品具有等效性。尽管如此,安全性和有效性是510(k)审查过程中满足等效性的基础。510(k)根据《联邦食品药品化妆品法案》第513(i)节[19]的规定来确定实质等效:待审批产品与对比产品具有相同预期用途和技术特征,或与对比产品具有相同预期用途,技术特征虽然不完全相同,但提交给FDA的信息能证明待审批产品与对比产品一样安全有效。通常,单一医疗器械不能用作医疗器械主导组合产品的对比产品。
4.2 药品主导型组合产品
新药申请(new drug application,NDA)和简略新药申请(abbreviated new drug application,ANDA)通常适用于药品主导型组合产品的上市申请。
4.2.1 新药申请
新药申请(NDA)是创新药品主导组合产品比较常见的申请途径。药品主导型组合产品的NDA必须包含证明产品在提议的使用说明中所规定、推荐或建议的内容的安全性和有效性。《联邦食品药品化妆品法案》第505节[20]描述了两种类型的NDA。505(b)(1)申请也称为独立 NDA,包含由申请人、或为申请人进行研究所获得的,或申请人有权参考或使用的安全性和有效性调查的完整报告。505(b)(2)申请同样需要包含有关安全性和有效性调查的完整报告,但有一些批准所需的安全性或有效性信息来自非申请人,或为非申请人进行的研究,并且申请人没有获得资料的参考权或使用权。
4.2.2 简略新药申请
仿制药主导的组合产品如果与已上市的对照药(reference listed drug,RLD)具有相同的活性成分、药物剂型、给药剂量和途径、使用条件以及使用说明(允许一定许可范围内的差异),则简略新药申请(ANDA)是适当的申请途径。ANDA必须包含足够的信息来证明待审批产品与RLD具有生物等效性,并且确保产品的特性、强度、质量和纯度。ANDA申请人无需提供独立证据来确定待审批产品的安全性和有效性,ANDA依赖于FDA对RLD安全有效的确认。药品主导的组合产品的ANDA应充分证明非主导成分与药品成分的最终配方兼容。虽然FDA并不期望待审批的仿制药组合产品与其RLD在所有方面都是相同的,但应充分分析待审批的仿制药组合产品与其 RLD之间的任何差异,并进行科学论证,否则可能无法获得ANDA的批准。
4.3 生物制品主导型组合产品
美国《公共卫生服务法》规定,生物制品主导型组合产品需要通过生物制品许可证申请(biologics license applications,BLA)的途径获得上市许可。BLA有两种:351(a)BLA(即“独立”BLA)和351(k)BLA(“生物类似药”或“可互换”生物制品BLA)[21]。
4.3.1 351(a)BLA
生物新药为主导的组合产品通常适用于351(a)BLA。这类产品主要包括:与基因疗法结合的特殊输送导管、预装注射器中的疫苗和自动注射器中的蛋白产品。BLA必须证明生物产品是安全、纯净和有效的,并且制造、加工、包装和保存生物产品的设施必须能达到确保生物产品保持安全、纯净和有效的标准。
4.3.2 351(k)BLA
351(k)BLA通常适用于以“生物类似药”或“可互换”生物制品为主导的组合产品。《公共卫生服务法》第351(k)节规定待审批产品应被证明与FDA批准的对比产品具有生物相似性或可互换性。生物相似性被定义为待审批产品与对比产品高度相似(尽管临床非活性成分存在微小差异),并且两种产品在安全性、纯度和效力方面没有临床意义的差异。为了满足互换性标准,申请人必须证明其产品与对比产品具有生物相似性,并且必须进一步证明该产品可以在任何给定患者中产生与对比产品相同的临床结果。对于向个人给药不止一次的产品,两种产品之间交替使用或转换使用的安全性和有效性降低的风险不大于没有这种交替或转换使用的情况。
关于组合产品生物相似性的证明,《生物药开发生物药价格竞争与创新法案》行业问答指南的Q.I.4指出,与待审批的生物类似药产品一起使用的输送装置的一些设计差异可能是允许的[22]。例如,对比产品是一种以瓶装形式获得批准的生物制品,而待审批的生物仿制药产品的输送装置是预装注射器或自动注射器,只要待审批产品符合法定生物相似性的标准,就是可以获得批准的。
5 展望
FDA对于现有组合产品的监管已经日臻成熟,但随着科学技术的发展,创新型组合产品不断刷新现有组合产品的概念,例如数字化药物,由算法控制的给药系统,带有活细胞的植入器械等,都将对监管提出新的挑战。随着组合产品复杂程度的提高,产品属性的界定也将保持其监管难点的地位。有研究人员提出在PMOA的基础上增加新的变量(主要使用意图,primary intended use,PIU)来提高产品属性的可预测性[23]。相信FDA会不断引入新的研究结果和技术,进一步提高监管的效率。FDA也一直鼓励组合产品的申请人在产品研发早期进行受理前的交流和讨论,以帮助申请人了解在申报准备中需要关注的问题,采用适当的研发策略。2021年全球医疗组合产品市场价值为1 181.3亿美元,预计到2030年市场规模将达到2 519亿美元,复合年增长率为 8.8%[24]。这是一个机遇与挑战并存的市场,相信在新技术的推动下,组合产品将在提高医疗效率和解决临床难题等方面发挥更大的作用。
猜你喜欢
猪业科学(2022年10期)2022-11-03
现代仪器与医疗(2022年2期)2022-08-11
吉林畜牧兽医(2022年7期)2022-07-20
猪业科学(2022年2期)2022-04-21
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2022年1期)2022-04-19
猪业科学(2022年1期)2022-03-24
现代仪器与医疗(2021年2期)2021-07-21
留学(2019年12期)2019-07-29
侨园(2018年7期)2018-10-09
