
[Ir(ppy)2(py-x)]+配合物的密度泛函理论研究
2022-08-10 03:13聂建航金雨廷张建坡
吉林化工学院学报 2022年3期
聂建航,王 哲,金雨廷,张建坡,金 丽
(吉林化工学院 化学与制药工程学院,吉林 吉林 132022)
在数十年的研究中,过渡金属配合物发光材料由于在OLED技术中的广泛应用[1-3],越来越多的OLED产品相继问世,特别是能提升发光效率、寿命以及拓展发光区域的新材料开发具有重要意义.而在一系列过渡金属中,磷光阳离子铱(Ш)配合物因其显示出覆盖整个光谱的可调激发和发射波长以及独特的光物理特征引起了社会的广泛关注,使其具有重要的科研价值和应用前景[4-5].
有研究表明,在激发态Ir与双(吡唑-1-基)甲烷的结合比主要的苯基吡啶配体弱得多,双(吡唑-1-基)甲烷的存在确保电子进一步定位在主要的苯基吡啶配体上,这导致三线态发射相对于以前使用芳香辅助配体的复合物显著蓝移[6].为了进一步探究含有双(吡唑-1-基)甲烷配体的Ir配合物的发光规律,选取一类[Ir(ppy)2(py-x)]+配合物采用量子计算方法进行理论探究,考察双(吡唑-1-基)甲烷配体上取代基的差异对分子结构和发光性质的影响.
1 计算方法
在曙光A620-G30服务器上使用Gaussian09程序包完成全部计算.使用密度泛函[7](Density functional theory)水平下的B3LYP[8]泛函和单激发组态相互作用(CIS)[9]方法分别优化了配合物基态和激发态几何结构,依据Franck-Condon垂直跃迁原理应用含时密度泛函[7]方法结合PCM[10](Polarized continuum modle)溶剂化模型计算了[Ir(ppy)2(py-x)+]配合物1-3在CH2Cl2溶液中的吸收和发射情况.对Ir原子使用只考虑价电子的赝势LANL2DZ[11]基组计算,对C、N、H原子使用6-31 G基组计算.该方法对于此类配合物的计算已被证实是可靠的.
2 结果和讨论
2.1 基态和激发态结构
图1为3个分子的基态结构图,相对应的基态和激发态几何参数还有实验值都在表1体现.从表1中可以看出,3个配合物都具有1A基态和3A激发态.配合物1-3的基态Ir-N(1)、Ir-N(4)、Ir-N(6)键长与实验值之差都在0.11Å范围内,键角N(1)-Ir-N(4)与实验值相差2.5 °,计算值和实验值的偏差处于合理范围内.激发态时,选择与Ir相连的4个主要键长都有所延长,表明激发态时电子跃迁由金属向配体跃迁,削弱了金属和配体之间的相互作用,且激发态时键角和二面角变化都不大.由表1中列出的3个分子主体结构数据可得出,吡咯、吡唑配体的引入并未对配合物主体结构造成较大影响.
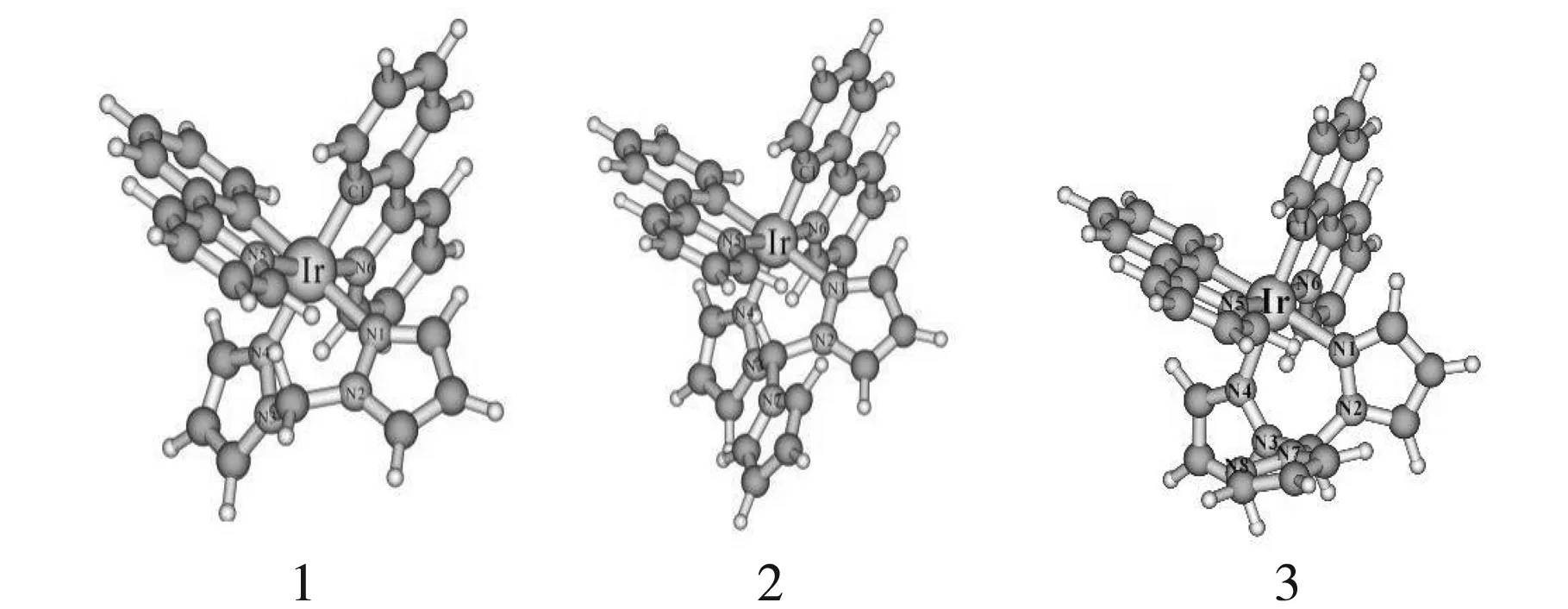
图1 配合物1-3的优化结构简图

表1 分别用B3LYP和CIS方法计算得到配合物1-3基态以及激发态的主要几何参数
2.2 配合物的吸收光谱
在CH2Cl2溶剂中,分别记录了分子1、2、3以TD-DFT(含时密度泛函)方法得到的Gaussian型基态吸收光谱如图2所示.

Wavelength/nm图2 配合物1-3在CH2Cl2溶液中模拟的Gaussian型吸收曲线
在表2中给出了选择的主要吸收和发射数据,包括跃迁轨道、波长、能量、振荡强度和所属性质.在表3中陈列出其提及的分子轨道相关成分和能量.

表2 配合物1-3在CH2Cl2溶剂中的吸收和发射光谱数据

表3 配合物1-3在B3LYP泛函水平下的基态分子轨道成分
从表2和图2中能够发现,3个分子都有两个高强度吸收带和1个强度较小最低能吸收带.配合物1-3的最低能吸收分别为399.07(3.11),401.68(3.09)和396.15(3.13) nm,配合物1的399.07 nm的吸收来自分子轨道128→129的激发,轨道128为最高占据轨道,主要由48.1%的d(Ir)和48.2%的π(ppy)构成,而129为最低空轨道,主要由90.3%的π*(ppy)配体占据.因此,该跃迁归属于d(Ir)+π(ppy)→π*(ppy)的金属到配体和ppy配体内部的电荷转移(MLCT/ILCT)跃迁.配合物2的401.68 nm的吸收也是来自HOMO到LUMO的跃迁,并且与配合物1有相似的跃迁性质,但配合物3略有不同,它的396.15 nm的吸收来自分子轨道145(HOMO)到146(LUMO)的激发,HOMO轨道主要由45.1%的d(Ir)和51.4%的π(ppy)构成,LUMO轨道由40.3%的π*(ppy)和54.1%的π*(py-pz)构成,因此该跃迁仅属于d(Ir)+π(ppy)→π*(ppy)+π*(py-pz)的金属到配体py-pz的跃迁,ppy配体成分在跃迁前后基本不变.通过分析3个配合物的最低能吸收波长可以发现,吡咯的引入,使配合物2的最低能吸收波长发生较小的红移(2.61 nm),吡唑的引入,使配合物3的最低能吸收波长发生较小的蓝移(2.92 nm).
相对于最低能吸收,其他两个吸收带明显增强.观察图2,第2吸收带分布在301 nm到327 nm之间的宽阔区域,第3吸收带为高能吸收带,振子强度最大,集中分布在260 nm到272 nm之间,波长相差较小.不同配体的引入对吸收带的位置产生了一定影响.根据上文表2、3可知,吸收带2分别由HOMO-2→LUMO、HOMO-4→LUMO+1、HOMO-3→LUMO+1贡献,且跃迁属性都归属于MLCT/ILCT跃迁.吸收带3分别由HOMO-3→LUMO+3、HOMO→LUMO+4、HOMO+4→LUMO+4激发捐献.以分子1为例,HOMO-3由41.8%金属Ir和49.7%的ppy配体π成键轨道占据,LUMO+3是由75.2%的py-H配体和20.5%的ppy配体构成的π反键轨道占据,经分析得出此跃迁被指认为MLCT/LLCT混合跃迁.同理分析得出配合物2、3在该位置拥有与配合物1相似的跃迁属性.
2.3 配合物的磷光发射光谱
配合物1-3在CH2Cl2溶液中的磷光光谱数据如表2所示,3个分子的磷光发射分别位于513.00 nm(1)、513.78 nm(2)、510.38 nm(3).他们都起源于HOMO→LUMO的激发,分子1、2的HOMO轨道主要占据在金属Ir和ppy配体上,而LUMO轨道只由ppy配体捐献,因此最低能吸收具有相似的跃迁属性,但配合物3略有不同,其LUMO由40.3%的py-pz配体和54.1%的ppy配体构成.比较配合物1-3的最低能吸收和发射(HOMO→LUMO),配合物1、2具有相似的轨道成分和跃迁性质(MLCT/ILCT),配合物3则因吡唑的引入略有不同(MLCT).能量差分别为0.69、0.68和0.70 eV,其斯托克斯频移非常接近,表明取代基的引入对此类配合物的影响较小.
3 结 论
本文从理论上对配合物[Ir(ppy)2(py-H)]+(1),[Ir(ppy)2(py-pr)]+(2),[Ir(ppy)2(py-pz)]+(3) 的基态以及激发态结构、轨道占据、光谱性质和跃迁属性进行了详细的研究.结果表明:HOMO轨道占据成分不受引入吡咯、吡唑配体的干扰,但LUMO轨道成分在引入吡唑配体后发生很大变化,分子1的LUMO主要为90.3%的π*(ppy),分子2的LUMO主要为89.3%的π*(ppy),而分子3的LUMO主要为54.1%的π*(ppy)和40.3%的π*(py-pz),从而使跃迁性质发生根本性的改变.吡咯和吡唑的引入都使HOMO和LUMO的轨道能和跃迁波长发生明显改变,吡咯的引入使分子2的吸收和发射红移,吡唑的引入使分子3的吸收和发射蓝移,但总体移动趋势都比较小,这主要是由于双(吡唑-1-基)甲烷配体本身为非共轭配体,在甲基部分引入取代基团依然不能产生共轭效应,因此对配合物的吸收和发射影响都不大,要想改变配合物的发光颜色需要围绕ppy配体来进行分子设计.
猜你喜欢
无线电工程(2022年10期)2022-10-24
数学物理学报(2022年5期)2022-10-09
化学工业与工程(2022年1期)2022-03-29
数学物理学报(2021年5期)2021-11-19
延边大学学报(自然科学版)(2021年1期)2021-04-27
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
化学与粘合(2020年4期)2020-09-11
化工管理(2020年8期)2020-06-02
生物工程学报(2020年1期)2020-03-12
