
光甘草定/环糊精固体包合物的制备和性质
2022-09-01 08:01姚培培樊金玲李德锋张晓宇任国艳
食品科学 2022年16期
姚培培,樊金玲,李德锋,张晓宇,任国艳,杜 琳
(河南科技大学食品与生物工程学院,河南 洛阳 471023)
光甘草定(glabridin,GLD)是光果甘草特有的疏水性异黄酮类化合物,含量为0.1%~0.3%,具有抗色素异常沉积、抗氧化、抗细胞增殖、抗炎、增强记忆力、抗骨质疏松和抗菌等多种生物活性。GLD难溶于水(7 μg/mL,25 ℃),导致其在体内胃肠道中的溶出率低、吸收和生物利用率差,在水溶性基质的食品和药品等相关领域的应用也因此受到极大的限制。因此,提高GLD在水中的溶解度是开发其潜在应用价值的关键所在。
环糊精(cyclodextrin,CD)是直链淀粉在由芽孢杆菌产生的葡萄糖基转移酶作用下生成的一系列环状碳水化合物的总称,最常见的是-、-、-CD,分别由6~8 个葡萄糖单元通过-1,4-糖苷键连接而成。CD各葡萄糖单元的2、3、6位羟基由不同的官能团取代,可得到一系列衍生物,如6-羟丙基--环糊精(6-hydroxypropyl-cyclodextrin,6-HP--CD)、2,6-二甲基--环糊精(2,6-di--methyl--cyclodextrin,2,6-M--CD)和2-磺丁基--环糊精(2-sulfobutyl--cyclodextrin,2-SBE--CD)等。CD及其衍生物均具有一个由亲水的外表面和相对疏水的中心空腔构成的圆筒式结构,这种独特的结构特性使CD能够通过非共价力(范德华力、静电相互作用和氢键)与多种化合物尤其是疏水性化合物相互作用,将后者包合在空腔中,形成主-客体包合物,从而提高难溶性化合物的溶解度,进而提高在体内的吸收及生物利用率。空腔大小和取代基的种类是影响CD及其衍生物对客体包合能力的重要因素。
本研究通过分子对接和相溶解度结合的方法筛选出适宜包合GLD的CD,并进行固体包合物的制备;考察不同干燥方法、不同投料比对固体包合物的包合率、载药量和溶解度的影响;采用扫描电子显微镜(scanning electron microscopy,SEM)法、差示扫描量热(differential scanning calorimetry,DSC)法、傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)法和分子对接技术对固体包合物的形貌、GLD的存在形式、GLD与2-SBE--CD的相互作用和空间构象等结构表征进行分析;并在此基础上进一步研究GLD/2-SBE--CD固体包合物的体外溶出特性及GLD/2-SBE--CD固体包合物对HepG-2细胞增殖的抑制作用。
1 材料与方法
1.1 材料与试剂
GLD 泌阳草木青生物科技有限公司;2-SBE--CD、6-HP--CD 湖北恒硕化工有限公司;-CD、-CD、-CD、2,6-M--CD、噻唑蓝(methylthiazolyl tetrazolium,MTT) 上海源叶生物科技有限公司;无水乙醇、乙腈、石油醚、二甲基亚砜(dimethyl sulfoxide,DMSO) 天津市德恩化学试剂有限公司;溴化钾 天津市科密欧化学试剂有限公司;铝坩埚上海菁仪化工材料有限公司;DMEM培养基 美国Fetal Bovine Serum生物科技有限公司;胎牛血清 江苏恩莫阿赛科技有限公司;胰消化酶 合肥Biosharp科技有限公司;HepG-2细胞 ATCC细胞库。
1.2 仪器与设备
SCIENTZ-10N冷冻干燥机 宁波新芝生物科技股份有限公司;pHS-3C pH计、L5S紫外分光光度计上海仪电科学仪器股份有限公司;TENSPOR27 FTIR仪德国Bruker仪器公司;DSC1型DSC仪 瑞士Mettler-Toledo公司;6000Y型喷雾干燥机 上海Bilon仪器有限公司;TM3030 SEM 日本日立高新技术公司;E191IR恒温培养箱 美国西蒙公司;CKX41SF倒置电子显微镜 日本Olympus公司;SW-CJ-2FD双人单面超净工作台 苏州净化设备有限公司;RS-232C酶标仪 美国Bio-Rad公司。
1.3 方法
1.3.1 CD的筛选
通过分子对接技术和相溶解度法测定不同种类CD与GLD的包合能力及稳定性,筛选适宜包合GLD的CD。
1.3.1.1 分子对接法
从PubChem数据库(https://pubchem.ncbi.nlm.nih.gov/)下载GLD的3D模型(CID编号为124052),用Gaussian 09软件中的DFT方法(B3LYP/6-31G)对3D模型进行优化。-CD、-CD和-CD的3D模型从剑桥晶体数据库(https://ccdc.cam.ac.uk/)得到,编号分别为1106001、1107194和1529141。用Gauss View 5.0软件打开去水后的-CD模型,用相应的取代基将葡萄糖单元中对应2,3,6位取代,得到-CD衍生物模型;其中,2,6-M--CD、单-6-氨基--CD、三乙酰基--CD的取代度分别为14、1、21,其他-CD衍生物取代度均为7;用Gaussian 09软件半经验算法中的PM6基组对-CD衍生物模型的几何构型进行优化。
采用AutoDockTools 4.2软件处理受体(-CD、-CD、-CD及-CD衍生物(6-HP--CD、2,6-M--CD、2-SBE--CD、6-硫酸盐--CD、单-6-氨基--CD、6-羧甲基--CD、三乙酰基--CD、6-季铵--CD))与配体(GLD),添加H原子与原子电荷,并设置GLD分子内可旋转单键数量及根原子。将设置好的受体和配体保存为pdbqt格式。在分子对接过程中,均以受体几何中心为中心,建立尺寸为60 Å×60 Å×60 Å的反应约束盒子。搜索参数选用拉马克遗传算法,算法对接的轮数设为100,能量评估的最大数目设为250 000,其他参数取默认值。对接方法采用半柔性对接。
1.3.1.2 相溶解度法
根据1.3.1.1节计算机模拟得到的结果选择恰当的-CD衍生物用于本实验。准确称取适量-CD、-CD、-CD及-CD衍生物,用去离子水配制浓度分别为0、10、20、30、40、50 mmol/L的CD溶液;其中,-CD和-CD溶解度较小,配制其溶液浓度为0、2、4、6、8、10 mmol/L。向上述不同浓度的CD溶液中加入过量的GLD,置于恒温振荡器中,25 ℃、200 r/min避光条件下孵育24 h。取出样品溶液,5 000 r/min离心5 min,取上清液。用80%乙醇溶液(/)适当稀释,在281 nm波长处测其吸光度,计算GLD的浓度。以CD浓度为横坐标,GLD浓度为纵坐标,绘制相溶解度图。
计算包合平衡常数()和包合效率(complexation efficiency,CE):

式(1)、(2)中:为相溶解度图直线斜率;为直线截距。
计算GLD和CD物质的量比(∶):

计算包合的吉布斯自由能(Δ):

式(4)中:R为气体常数,数值为8.314 J/(mol·K);为开氏温度/K。
1.3.2 GLD/CD固体包合物的干法制备
采用捏合法制备固体包合物。将GLD与筛选出最适宜的CD按物质的量比1∶1称量。用2 倍CD质量的去离子水溶解CD,研磨均匀。用无水乙醇溶液溶解GLD,配制成30 mg/mL的GLD乙醇溶液。将GLD乙醇溶液全部滴加到CD溶液中,研磨45 min,60 ℃烘箱中干燥至质量恒定,得到固体包合物。
1.3.3 GLD/CD固体包合物的湿法制备
将GLD与筛选出最适宜的CD按的物质的量比1∶1称量。用去离子水溶解CD,配制成0.04 mol/L的CD溶液。用少量无水乙醇溶解GLD,配制成30 mg/mL的GLD乙醇溶液。将GLD乙醇溶液加入到CD溶液中,使体系中乙醇体积分数为30%,于25 ℃、200 r/min的条件下振荡24 h。分别将所得溶液用冷冻干燥法、喷雾干燥法和共蒸发干燥法进行干燥,制备固体包合物。
将GLD与CD按物质的量比1∶1.5和1.5∶1称量,重复上述操作,经冷冻干燥后,得到不同投料比的固体包合物。
1.3.3.1 冷冻干燥法
用45 ℃的旋转蒸发器除去所得溶液中的乙醇,将溶液置于-20 ℃冰箱中预冻12 h。置于冷冻干燥机中冻干后,过80 目筛。
1.3.3.2 喷雾干燥法
用45 ℃的旋转蒸发器除去所得溶液中的乙醇。将溶液置于喷雾干燥器中干燥,采用1 mm的加压雾化器,进样速度2.3 mL/min,进风温度180 ℃,排风温度110 ℃,雾化气流速度3.9 m/min。
1.3.3.3 共蒸发干燥法
将所得溶液置于60 ℃的旋转蒸发器中旋转蒸发至干。
1.3.4 GLD/CD固体包合物饱和溶解度的测定
分别在1 mL去离子水中加入过量的经不同制备方法得到的GLD/CD固体包合物,在室温条件下超声溶解至平衡状态。0.45 μm滤膜过滤,用80%乙醇溶液逐步稀释滤液到合适浓度(稀释10 000 倍)。在281 nm波长处测定吸光度,计算GLD的含量,即为饱和溶解度。
1.3.5 GLD/CD固体包合物包合率和载药量的测定
分别称取两份10 mg经不同制备方法得到和不同投料比的GLD/CD固体包合物。一份溶于1 mL去离子水中,加入9 mL乙腈25 ℃超声处理20 min。5 000 r/min离心5 min后,收集上清液,用乙腈稀释10 倍。测定其在281 nm波长处的吸光度,根据标准曲线计算GLD的质量,即为包合物样品中GLD总质量。
另一份加入400 μL石油醚,充分混匀。5 000 r/min离心5 min,去上清液(用来洗去未被包合的GLD),重复洗2 次,置于烘箱中使石油醚挥发。按上述包合物样品中GLD总质量的测定步骤测定此样品中的GLD质量,即被包合的GLD质量。计算包合率和载药量:

1.3.6 GLD/CD固体包合物及相关样品的结构表征
1.3.6.1 SEM分析
对GLD、CD、GLD/CD物理混合物(以质量比1∶100混合均匀)及经不同制备方法得到的GLD/CD固体包合物进行扫描电子显微镜测试。用导电双面胶将样品固定在样品台上,喷镀铂金后在3 kW条件下,分别于300、400、1 200 倍观察样品的表面形态。
1.3.6.2 DSC分析
分别称取GLD、CD、GLD/CD物理混合物及经不同制备方法得到的GLD/CD固体包合物各5 mg,温度扫描范围25~300 ℃,升温速率10 ℃/min,氮气流量10 mL/min,以空盘作为参比,记录各样品的DSC图线。
1.3.6.3 FTIR分析
分别称取适量的GLD、CD、经不同制备方法得到的GLD/CD固体包合物和GLD/CD物理混合物,分别与溴化钾粉末混合,放入研钵内充分研磨,再采用压片法制得样品薄片。将样品薄片放入仪器内进行光谱扫描,扫描范围4 000~400 cm,分辨率4 cm,扫描次数32 次。以纯溴化钾粉末作背景进行单通道扫描。
1.3.6.4 分子对接法
采用1.3.1.1节的步骤,运用AutoDockTools 4.2软件对GLD与筛选出最适宜的CD包合模式进行模拟得到主客体间的构象及氢键相互作用。
1.3.7 GLD/CD固体包合物在模拟胃肠液中累积溶出率测定
按照Maltais等所述方法配制不含消化酶的模拟胃、肠液。准确称取2.0 g NaCl置于烧杯中,加入100 mL去离子水,用12.0 mol/L浓盐酸调节pH值至1.2,即为模拟胃液。准确称取6.8 g KHPO,溶于250 mL去离子水中,用0.2 mol/L NaOH溶液调节pH值至6.9,即为模拟肠液。分别量取900 mL模拟胃液和肠液作为溶出介质,放入1 000 mL烧杯中,置于水浴加热磁力搅拌器,温度保持在(37±0.5) ℃,转速50 r/min。分别准确称取一定质量的GLD、GLD/CD物理混合物及经不同制备方法得到的GLD/CD固体包合物样品(样品中GLD质量均为100 mg),放入烧杯中,并立即开始计时。于2、4、6、8、10、15、20、30、45、60 min时吸取溶出液1 mL,同时补充等体积新鲜的溶出介质。用0.45 μm微孔滤膜过滤溶出液,取滤液0.5 mL,加入4.5 mL乙腈,充分混匀。5 000 r/min离心5 min后,取上清液。在281 nm波长处测定吸光度,计算累积溶出率:
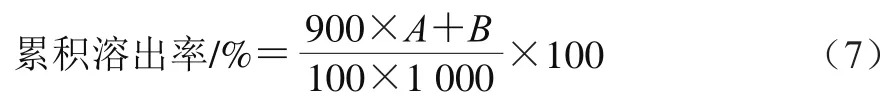
式(7)中:为某一时间点每1 mL溶出液中溶出的GLD质量/μg;为之前所有时间点取出的1 mL溶出液中GLD质量/μg。
绘制溶出曲线,按下式计算差异因子()和相似因子(),进行相同条件下各溶出曲线间的差异显著性分析。

式(8)、(9)中:为取出点的数量;R为参考曲线在时间点的溶解率;T为比较曲线在时间点的溶出率。
当=0、=100,表明测试曲线和参考曲线完全相似;当值在0~15内且值在50~100内,表明两条曲线相似;当>15且<50时,表明两条曲线差异显著;其余情况无法判定曲线间的差异显著性。
1.3.8 GLD/CD固体包合物对HepG-2细胞增殖的抑制率测定
1.3.8.1 样品制备
样品组中GLD、CD和GLD/CD的系列浓度均为30、60、90、120、150、200 μmol/L。将GLD溶于DMSO中,配制50 mmol/L母液,用DMEM培养基稀释得到不同浓度的GLD样品,记为GLD/DMSO组。将GLD溶于DMEM培养基中得到不同浓度的GLD样品,记为GLD/HO组。将筛选出最适宜的CD溶于DMEM培养基中得到不同浓度的CD样品,记为CD组。将经冷冻干燥得到的GLD/CD固体包合物复溶于DMEM培养基中得到不同浓度的GLD/CD样品,记为GLD/CD组。
1.3.8.2 MTT法测定HepG-2细胞增殖的抑制率
将含体积分数10%的胎牛血清、100 U/mL双抗的DMEM培养基置于37 ℃、含5% CO气体的恒温培养箱中,培养HepG-2细胞至80%的细胞铺满瓶底。用胰酶消化2 min后,以1×10个/mL的浓度接种至96 孔板,每孔200 μL,培养16 h。弃去旧培养基,加入200 μL新鲜培养基或样品液,继续培养24 h。弃去旧培养基,用磷酸盐缓冲液清洗两次,每孔加入200 μL新鲜培养基和10 μL 5 mg/mL MTT溶液,继续培养4 h。倒掉培养基中液体,吸掉泡沫,加150 μL DMSO反应10 min。用酶标仪测定550 nm波长处样品孔的吸光度。加MTT的样品孔记为,加等体积培养基的孔记为,不加MTT的孔为记,每个样品做6 个复孔,按式(10)计算细胞抑制率,并根据浓度-细胞抑制率曲线求得半抑制浓度(half maximal inhibitory concentration,IC)。
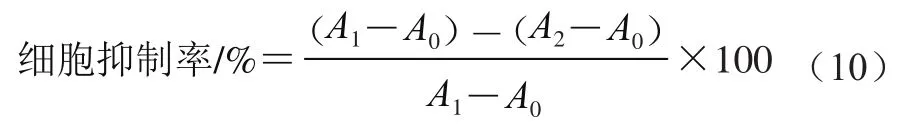
1.4 数据处理
除说明外,其他所有实验重复3 次,采用DPS软件进行差异显著性分析,采用Origin 8.5软件作图。
2 结果与分析
2.1 CD与GLD的包合能力及包合物的稳定性分析
2.1.1 分子对接法筛选CD的结果分析
不同种类CD与GLD对接的最佳结合能见表1。-CD和-CD最佳结合能低于-CD,说明与-CD相比,-CD、-CD与GLD可形成更加稳定的包合物。在-CD的衍生物中,三乙酰基--CD和6-季铵--CD这两种-CD衍生物经取代后与GLD的最佳结合能均高于-CD;2-SBE--CD、6-HP--CD、2,6-M--CD、6-硫酸盐--CD、单-6-氨基--CD和6-羧甲基--CD经取代后与GLD的最佳结合能均低于-CD。其中,2-SBE--CD与GLD对接的结合能明显低于其他种类CD,为-8.60 kcal/mol。这些结果说明,CD空腔大小和取代基的种类是影响CD及其衍生物对客体包合能力的重要因素,2-SBE--CD与GLD包合的稳定性最好。
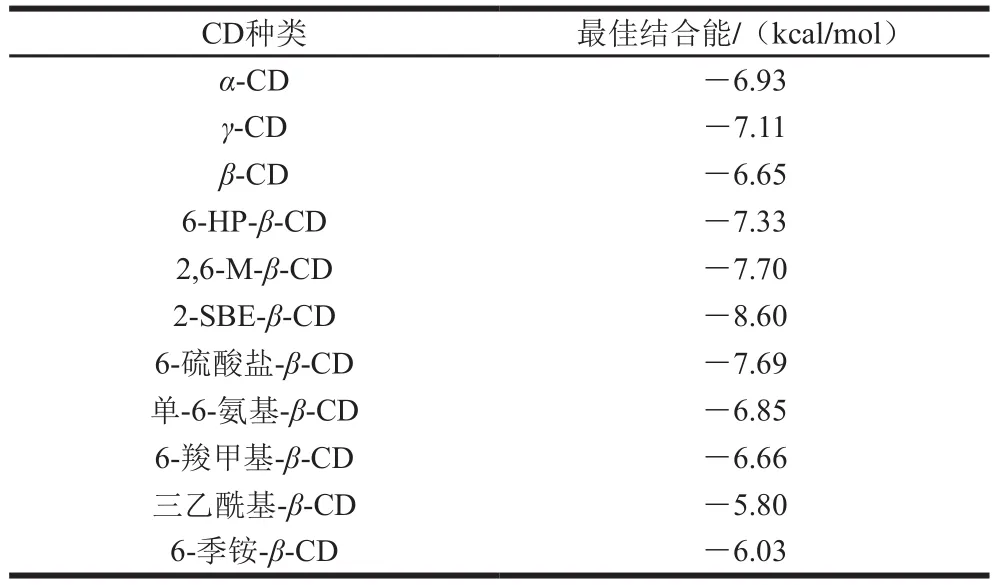
表1 不同种类CD与GLD的最佳结合能Table 1 Optimal binding energy between different CDs and GLD
2.1.2 相溶解度法筛选CD的结果分析
选取-CD、-CD、-CD及3 种结合能较小的-CD衍生物(6-HP--CD、2,6-M--CD和2-SBE--CD),采用相溶解度法进一步研究其与GLD的包合作用。其中,-CD未能与GLD形成包合物,其他5 种CD与GLD包合的相溶解度曲线如图1所示。所有曲线线性回归方程的相关系数()均大于0.98(表2),即相溶解度曲线为A型,表明CD与GLD以1∶1物质的量比进行包合。值与包合物稳定性有关,值越大,表明GLD与CD形成的包合物稳定性越好。由表2可知,-CD以及3 种-CD衍生物的均大于-CD,表明其与GLD的包合稳定性均优于-CD;其中,2-SBE--CD与GLD包合的最大,与GLD形成的包合物最稳定。GLD与不同CD包合过程中的Δ均为负值,表明包合过程在25 ℃时均可自发进行;-CD以及3 种-CD衍生物的Δ均低于-CD,2-SBE--CD与GLD包合的Δ低于其他CD,与上述分子对接的结合能结果吻合。

图1 不同种类CD与GLD的相溶解度曲线Fig. 1 Phase solubility of different CDs and GLD

表2 CD和GLD包合的拟合方程、K1∶1、CE及热力学相关参数Table 2 Fitting equations, K1:1, CE and thermodynamic parameters for inclusion complexation between CD and GLD
对于难溶性药物或生物活性成分(水溶性<0.1 mmol/L),其固有溶解度通常远小于相溶解度曲线的,导致值计算误差较大。CE值越大,∶值越大,表明主体分子对客体分子的包合能力更强,增溶能力越好;CE和∶值的大小与客体分子的固有溶解度(或)无关,因此可以更准确地反映包合效果。从表2可知,与-CD相比,3 种-CD衍生物的CE和∶均增大,表明-CD衍生物与GLD的包合能力均优于-CD;其中,2-SBE--CD与GLD包合的CE和∶值最高,说明其包合能力最强。
上述结果可以看出,通过分子对接模拟技术发现2-SBE--CD与GLD对接的结合能最低、最稳定;相溶解度实验证明2-SBE--CD对GLD的包合效果最好;因此,选取2-SBE--CD进行后续实验。
2.2 GLD/2-SBE-β-CD固体包合物的包合率、载药量及饱和溶解度分析
2.2.1 制备方法对GLD/2-SBE--CD固体包合物的影响

图2 冷冻干燥法(a)、捏合法(b)、喷雾干燥法(c)及共蒸发法(d)制得的GLD/2-SBE-β-CD固体包合物Fig. 2 Pictures of GLD/2-SBE-β-CD solid inclusion complexes prepared by freeze drying (a), kneading (b), spray drying (c) and co-evaporation (d)
GLD与2-SBE--CD物质的量比为1∶1时,经干法制备和3 种湿法制备方法得到的GLD/2-SBE--CD固体包合物如图2所示。由表3可知,4 种制备方法所得包合物的包合率和载药量差异不显著,包合率为93.33%~97.68%,载药量为11.19%~11.89%;不同干燥法制备的包合物能够明显提高GLD的饱和溶解度(未经包合的GLD饱和溶解度为7 μg/mL),这是因为CD具有一个亲水的表面结构,其空腔内壁由碳链骨架构成,表现出疏水性,因此可以和难溶性化合物通过范德华力或氢键作用形成包合物,从而提高难溶性化合物的溶解度。除了共蒸发法制得的包合物的饱和溶解度显著低于喷雾干燥法外,其他3 种制备方法(冷冻干燥法、喷雾干燥法和捏合法)制得的包合物的饱和溶解度差异不显著,均大于83 mg/mL。

表3 不同干燥方法制得的GLD/2-SBE-β-CD固体包合物的包合率和载药量Table 3 Inclusion rates and drug-loading rates of GLD/2-SBE-β-CD solid inclusion complexes prepared by different drying methods
2.2.2 投料比对GLD/2-SBE--CD固体包合物的影响
由表4可知,伴随GLD与2-SBE--CD物质的量比增大,包合率下降、载药量提高。GLD与2-SBE--CD物质的量比为1.5∶1时,包合率为86.09%,载药量为22.39%,与GLD和2-SBE--CD物质的量比为1∶1、1∶1.5相比,包合率分别下降了7.75%、8.58%,载药量则分别提高了88.31%、141.14%。

表4 不同投料比制得的GLD/2-SBE-β-CD固体包合物的包合率和载药量Table 4 Inclusion rates and drug-loading rates of GLD/2-SBE-β-CD solid inclusion complexes prepared with different GLD/2-SBE-β-CD ratios
上述研究结果表明:不同制备方法对GLD/2-SBE--CD固体包合物的包合率和载药量均无显著影响,但对包合物的水溶性有一定影响。适当提高GLD与2-SBE--CD物质的量比,包合率虽有一定程度下降,但可显著提高载药量。
2.3 GLD/2-SBE-β-CD固体包合物的表征
2.3.1 SEM结果分析
如图3所示,2-SBE--CD为大小不一、表面光滑并有凹陷的球状结构,与文献报道相符;GLD呈柱状晶体结构,轮廓清晰;GLD/2-SBE--CD物理混合物中可同时观察到球状2-SBE--CD和柱状的GLD,表明是两者的简单混合。GLD/2-SBE--CD包合物的形貌特征明显不同于物理混合物,表明主客体分子之间发生了相互作用;且制备方法不同,包合物的外部形貌有较大差异:冷冻干燥法制备的包合物呈边缘锋利的片状结构;捏合法和共蒸发法制备的包合物为不规则的块状结构;喷雾干燥法制备的包合物为表面光滑、粒径很小的球形颗粒(小于10 μm),颗粒尺寸显著小于冷冻干燥法、捏合法和共蒸发法制得的包合物。

图3 2-SBE-β-CD(a)、GLD(b)、GLD/2-SBE-β-CD物理混合物(c)及不同制备方法得到的GLD/2-SBE-β-CD固体包合物(d~g)的SEM图Fig. 3 SEM images of 2-SBE-β-CD (a), GLD (b), GLD/2-SBE-β-CD physical mixture (c) and GLD/2-SBE-β-CD solid inclusion complexes (d–g)obtained by different drying methods
2.3.2 DSC结果分析
如图4所示,GLD在233 ℃处有尖锐的熔融峰,此温度是该晶体的熔点。2-SBE--CD有两个峰,在40~160 ℃内出现一矮而宽的吸热峰,为水由其空腔内释放的吸热峰;在270 ℃有一个吸热峰,此峰代表2-SBE--CD分解,这与文献报道的结果一致。GLD/2-SBE--CD物理混合物在233 ℃和270 ℃处均有吸热峰,体现为GLD与2-SBE--CD吸热峰的简单叠加,表明GLD在物理混合物中仍表现出晶体特征。通过冷冻干燥法、喷雾干燥法、捏合法和共蒸发法制得的GLD/2-SBE-CD固体包合物DSC图无明显差别(图中d曲线是经冷冻干燥法制备并测定所得,其余略)。与2-SBE--CD相比,GLD/2-SBE-CD固体包合物的第1个峰的峰强度降低且发生了偏移,表明将GLD插入空腔中会使水分子发生迁移,偏移的发生推测是GLD与2-SBE--CD之间形成了相互作用的氢键;第2个峰位偏移至276 ℃,对应于GLD/2-SBE-CD固体包合物中2-SBE--CD的分解;包合物的DSC曲线中无GLD的熔融峰,表明由于GLD/2-SBE-CD固体包合物的形成,晶态的GLD转变为非晶态。上述结果均说明了2-SBE--CD已将GLD包合在空腔内形成了固体包合物。

图4 GLD、2-SBE-β-CD、GLD/2-SBE-β-CD物理混合物和经冷冻干燥法得到的GLD/2-SBE-β-CD固体包合物的DSC分析图Fig. 4 DSC analysis of GLD, 2-SBE-β-CD, GLD/2-SBE-β-CD physical mixture and GLD/2-SBE-β-CD solid inclusion complex obtained by freeze-drying method
2.3.3 FTIR结果分析
如图5所示,在GLD的光谱中,3 346 cm波数处出现一强而宽的峰,为O—H的伸缩振动;1 518、1 464 cm波数处为芳香环C=C的伸缩振动;2 964、2 920 cm波数处为—CH的吸收峰。在2-SBE--CD的光谱中,3 430 cm波数前后表现出与O—H伸缩振动相关的特征波段;2 935 cm波数处为C—H的伸缩振动;1 653 cm波数处为水分子的弯曲振动;1 163、1 039 cm波数处为C—O的伸缩振动。在GLD/2-SBE--CD物理混合物的光谱中,GLD和2-SBE--CD的特征吸收峰依旧存在,表明物理混合物只是主客分子的简单叠加,GLD和2-SBE--CD之间没有相互作用或仅有微弱的相互作用。不同方法制备的GLD/2-SBE--CD固体包合物的FTIR图无明显差别(图中d曲线是经冷冻干燥法制备并测定所得,其余略)。在GLD/2-SBE--CD固体包合物中,GLD芳香环对应的特征吸收峰消失;2-SBE--CD中3 430 cm波数处O—H的吸收峰在包合物中移至3 407 cm波数处,且吸收带强度增加,这可能是由于GLD和2-SBE--CD之间发生了相互作用导致羟基数量增加;2-SBE-CD中1 653 cm波数处水分子的特征弯曲振动吸收峰在包合物中移至1 645 cm波数处。基于GLD/2-SBE--CD固体包合物FTIR图的变化,说明GLD进入了2-SBE--CD的空腔。GLD/2-SBE--CD固体包合物光谱中未出现新的吸收峰,表明包合物中无新的化学键产生,主客分子间只是分子间力或氢键的相互作用。

图5 GLD、2-SBE-β-CD、GLD/2-SBE-β-CD物理混合物和经冷冻干燥法得到的GLD/2-SBE-β-CD固体包合物的FTIR分析图Fig. 5 FTIR analysis of GLD, 2-SBE-β-CD, GLD/2-SBE-β-CD physical mixture and GLD/2-SBE-β-CD solid inclusion complex obtained by freeze-drying method
2.3.4 GLD与2-SBE--CD分子对接结果分析
如图6所示,GLD分子能够完整地进入2-SBE--CD的空腔内。GLD与2-SBE--CD分子间形成两个氢键:2-SBE--CD某两个相邻葡萄糖单元中C2位磺酸基的氧分别与GLD分子B环上C2位和C3位O—H的氢之间形成氢键;氢键距离分别为2.1、2.3 Å。表明氢键是GLD与2-SBE--CD相互作用的主要作用力之一。

图6 GLD与2-SBE-β-CD对接的最佳能量构象及相互作用图Fig. 6 Lowest energy conformation and interaction diagrams for GLD docking with 2-SBE-β-CD
2.4 GLD/2-SBE-β-CD固体包合物在模拟胃肠液中累积溶出率分析
难溶性的药物或生物活性成分的累积溶出率低,是导致其在体内的吸收能力低、生物利用率差的一个重要原因。因此,累积溶出率可以在一定程度上预测难溶性化合物体内的吸收速度和程度。不同制备方法得到的GLD/2-SBE--CD固体包合物的累积溶出率无显著差异,图7中曲线是经喷雾干燥法制备并测定所得,其余略。由图7可知,在pH 1.2的模拟胃液和pH 6.9的模拟肠液中,GLD及GLD/2-SBE--CD物理混合物在1 h的累积溶出率均小于10%;与之相比,GLD/2-SBE--CD包合物在模拟胃液和肠液中的累积溶出率均极显著提高,1 h的累积溶出率分别达到87%和83%。结果表明GLD/2-SBE--CD固体包合物能有效提高GLD的溶出,这是由于GLD与CD间的氢键相互作用以及GLD在包合物中的结晶度减少引起。这预示包合物具有提高GLD吸收的潜在能力。
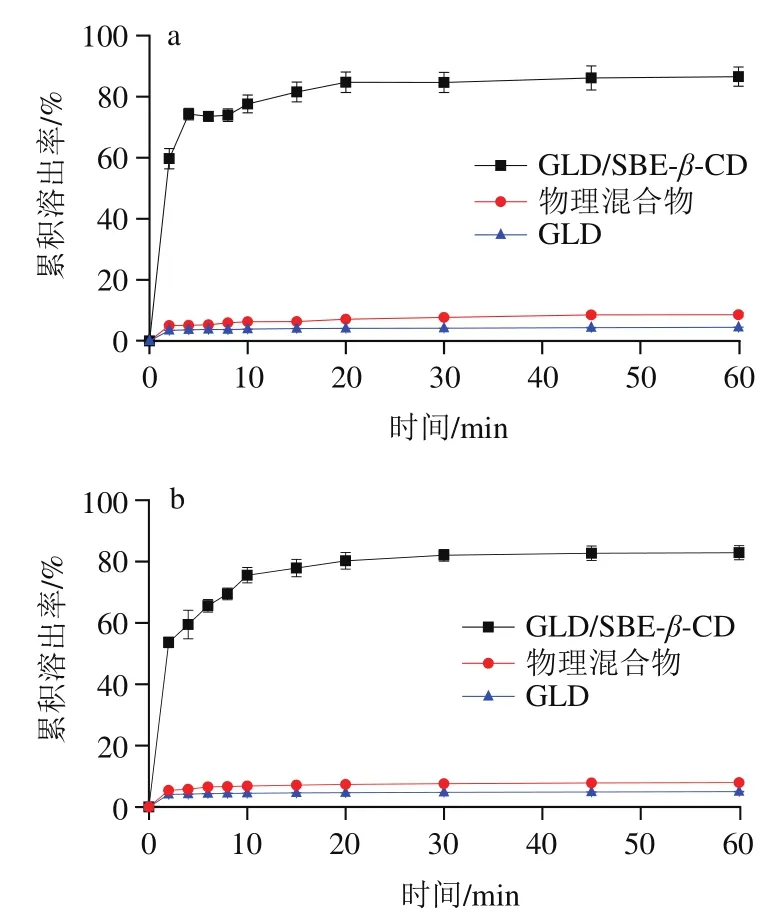
图7 经喷雾干燥法制备的GLD/2-SBE-β-CD固体包合物在模拟胃液(a)、肠液(b)中的累积溶出率Fig. 7 Cumulative dissolution rates of GLD/2-SBE-β-CD solid inclusion complex prepared by spray drying method in simulated gastric juice (a) and intestinal juice (b)
2.5 GLD/2-SBE-β-CD固体包合物对HepG-2细胞增殖的抑制作用
GLD具有抑制人乳腺癌细胞系(MCF-7、MDAMB-231和T-44D)、胚胎肾细胞HEK-293、血癌细胞K562、宫颈癌Hela、肝癌HepG-2、肝癌WRL-68、口腔癌SCC-9和肺癌细胞A546等多种癌细胞增殖的作用。本实验比较了GLD/HO组、GLD/DMSO组、2-SBE--CD组和经冷冻干燥法制得的GLD/2-SBE--CD组对HepG-2细胞增殖的抑制作用,结果如图8所示。GLD/DMSO组对HepG-2细胞增殖表现出很强的抑制作用,其IC为37.68 μmol/L。GLD/HO组对HepG-2细胞增殖的抑制作用较弱,其IC为162.29 μmol/L。2-SBE--CD组对HepG-2细胞增殖的抑制率均小于2%,表明2-SBE--CD几乎是无毒的。GLD/2-SBE--CD组对HepG-2细胞增殖抑制作用的IC值为74.69 μmol/L,显著优于GLD/HO组。
GLD在水中的溶解度低,导致GLD/HO处理对HepG-2细胞增殖的抑制作用低。DMSO起到增加GLD溶解性的作用,可以提供更多的游离GLD分子与HepG-2细胞接触,因此GLD/DMSO处理提高了对HepG-2细胞增殖的抑制作用。但因溶剂中含有DMSO,所以不能完全反映出游离GLD在水体系中的细胞抑制效果。GLD/2-SBE--CD固体包合物对HepG-2细胞增殖活性的抑制作用显著优于GLD/HO处理,可能的原因有:GLD/2-SBE--CD固体包合物的水溶性好,使更多溶解的GLD分子作用于细胞;GLD/2-SBE-CD固体包合物使GLD通过细胞膜的运输增加,从而提高了对细胞增殖的抑制活性。

图8 GLD/H2O组、GLD/DMSO组、2-SBE-β-CD组和经冷冻干燥法制得的GLD/2-SBE-β-CD组对HepG-2细胞增殖的抑制Fig. 8 Inhibitory effects of GLD/ H2O, GLD/DMSO, 2-SBE-β-CD and GLD/2-SBE-β-CD prepared by freeze-drying method on the proliferation of HepG-2 cells
3 结 论
除-CD外,-CD、-CD、6-HP--CD、2,6-M--CD和2-SBE--CD均可以不同程度地提高GLD的溶解度,与GLD包合的相溶解度曲线均为A型,表明与GLD形成物质的量比1∶1的包合物。其中2-SBE--CD包合GLD的能力优于其他CD及其衍生物;不同制备方法制备的GLD/2-SBE--CD固体包合物的包合率和载药量均无显著差异,但包合物的饱和溶解度有一定差异。适当提高GLD与2-SBE--CD物质的量比,包合率虽有一定程度下降,但可显著提高载药量。GLD与2-SBE--CD物质的量比为1.5∶1时,冷冻干燥法制备的GLD/2-SBE--CD固体包合物的包合率和载药量分别为86.09%和22.39%。GLD与2-SBE--CD物质的量比为1∶1时,冷冻干燥法、喷雾干燥法和捏合法制得的包合物的饱和溶解度均大于83 mg/mL。不同制备方法得到的GLD/2-SBE--CD包合物形貌差异明显,GLD在GLD/2-SBE--CD包合物中均以无定形非晶体结构存在。GLD/2-SBE--CD包合物能有效提高GLD在模拟胃、肠液中的溶出;制备方法对GLD/2-SBE--CD包合物的溶出特性无显著影响;同时,GLD/2-SBE--CD包合物还有效保证GLD的抗肿瘤增殖活性。因此,GLD/2-SBE--CD包合物是GLD的一种有前途的给药形式,值得深入开展药代动力学以及跨膜转运机制的相关研究,从而更好地扩大GLD的应用潜力。
猜你喜欢
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
科技视界(2018年18期)2018-11-09
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
学苑创造·B版(2016年5期)2016-07-18
中学生数理化·中考版(2015年11期)2015-09-10
中学化学(2015年5期)2015-07-13
老友(2010年3期)2010-03-25
