
不对称[5+1]串联合成螺[环己酮-氧化吲哚]衍生物
2022-10-18 05:08杨兰西王金娟陈治明
江西师范大学学报(自然科学版) 2022年3期
杨兰西,李 淼,王金娟,陈治明*
(1.贵州师范大学化学与材料科学学院,贵州 贵阳 550001;2.贵州师范大学贵州省功能材料化学重点实验室,贵州 贵阳 550001)
0 引言
螺氧化吲哚是在众多生物碱、天然产物和药物中最主要和最多样的框架之一[1],鉴于螺旋-吲哚酮显著的结构复杂性、有趣的生物学特性,其在对映选择性构建方面所面临的挑战引起了化学家和生物学家的关注[2-3].功能化的螺旋化吲哚杂合体显示出各种药用特性[4],该特性与天然生物碱的外观或相似性有关,使其在抗菌、抗癌、抗病毒药物等许多治疗领域中具有广泛的活性[5-7].尤其是具有手性螺旋[环己酮-吲哚啉酮]核心的螺环氧化吲哚家族,它是一个多立体环己酮和吲哚啉酮基序的有趣组合和颇具潜力的生物活性分子[8],曾经作为抗癌药、丙型肝炎抑制剂和孕酮受体调节剂获得多项专利[9].
目前报道的手性螺环己酮吲哚啉酮结构的构建方法有分子内环化、[4+2]环加成和[5+1]螺旋化3种环化模式.Zhang Yafei等[10]和S. Katayama等[11]分别报道了采用氧化吲哚衍生的二酮分子内Aldol反应和使用N-芳基-N-甲基-2-氧环己烷-1-羧基酰胺分子内环化制得螺旋环己酮吲哚啉酮;该策略要求反应底物的结构较为复杂,且反应的条件也要求极高.[4+2]环加成是合成螺环己烷吲哚二酮的最常见的方法,3-亚甲基吲哚酮是该策略的主要反应物.它是利用3-亚甲基吲哚酮存在外环缺电子的C3烯基键和Michael受体性质,使其在环加成或串联反应中表现为十分出色的合成子[12].G. Melchiorre等[13]通过[4+2]双Michael加成合成了手性螺旋[环己酮-吲哚啉酮],Wei Qiang等[14]、Lan Yubao等[15]、Zheng Jianfeng等[16]、Luo Yao等[17]也分别报道了用不同的[4+2]不对称环化方法合成螺环己烷吲哚啉二酮.[5+1]螺旋化也是合成螺环己酮氧化吲哚的一种重要策略,但相比于[4+2]环化,不对称[5+1]环化的文献报道较少[18].
本文以潜在的亲电受体二苯乙烯甲酮与吲哚啉酮为底物,以手性酰基胍为催化剂,利用氢键的催化能力有效地将2种羰基化合物的催化剂活化模式结合成一种机制,直接用一锅法合成螺氧吲哚环己酮衍生物.
1 实验部分
1.1 仪器和试剂
实验所用的仪器有:RE-52旋转蒸发仪(上海泸西分析仪器厂有限公司),DRX型400MHz核磁共振仪、UHR TOF LC型超高分辨飞行时间质谱(德国Bruker公司),Perkin Elmer Frontier型红外光谱仪、WZZ-1型旋光仪(美国Perkin Elmer公司),X-6型熔点测定仪(北京泰克仪器有限公司),RE53-3型气相色谱仪(上海天美有限公司).实验所用试剂均为市售分析纯级.
1.2 胍-酰胺催化剂1a~1c的合成
在50 mL反应瓶中加入2.5 mmol R型联二萘酚酸、4.2 mmol DCC和HOBt,然后加入适量THF使其溶解,缓慢滴加5.0 mmol胍基,在室温、氮气保护下反应,TLC 追踪反应至结束.反应混合液用CH2Cl2萃取3次,有机层用流动相为V(石油醚)∶V(乙酸乙酯)=10∶1的硅胶柱层析,纯化得到胍-酰胺催化剂1a~1c,合成路线如图1所示.

图1 催化剂1a~1c的合成
1.3 手性螺[环己酮-吲哚啉酮]化合物4a~4o的合成
将0.6 mmol的二苯乙烯基甲酮(2)和0.3 mmol的吲哚啉酮(3)依次加入25 mL的圆底烧瓶中,然后加入2 mL的二氯甲烷,在底物溶解后再加入0.006 mmol的催化剂1,在40 ℃的条件下搅拌反应72 h,用TLC跟踪反应至结束.将反应得到的产品用10 mL乙酸乙酯萃取2~3次,有机相用饱和氯化钠溶液洗剂后用无水硫酸镁干燥,在过滤、减压蒸馏除去溶剂后用柱层析(洗脱剂V(石油醚)∶V(乙酸乙酯)=6∶1)纯化,得到白色固体产物4a~4o, 合成路线如图2所示.
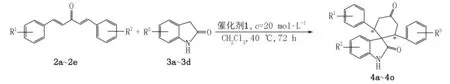
图2 目标产物4a~4o的合成
2 结果与讨论
2.1 催化剂及用量的筛选
以二苯乙烯基甲酮2a、吲哚啉酮3a为底物,以CH2Cl2为溶剂,在40 ℃下反应72 h,考察了3种结构不同的酰基胍催化剂的催化性能和最佳用量(见表1).
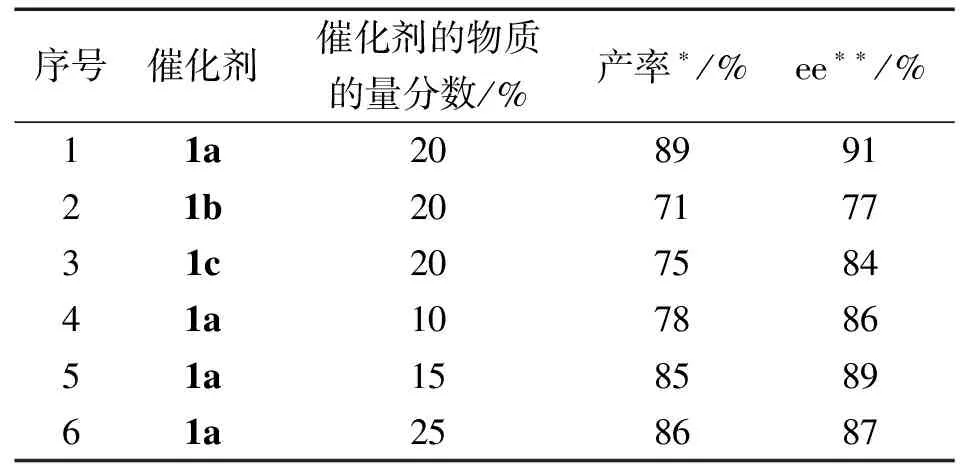
表1 二烯酮2a和吲哚啉酮3a的不对称[5+1]串联的催化剂及其用量研究
由表1可知:3种氢键型有机催化剂手性酰基胍都能有效催化二苯乙烯甲酮2a和吲哚啉酮3a的[5+1]环加成反应,单苯基取代的手性双胍1a能以89%的产率和91%的ee值获得螺环己酮吲哚啉酮衍生物,甲氧基取代的手性单胍1b和二苯基取代的双胍1c也能分别以71%、75%的产率和77%、84%的ee值获得目标产物4a.催化剂1a的苯环与胍基上的氮原子共轭增强了胍基上的氢与底物形成氢键的能力,同时手性胍上氢键的数目比叔丁基取代的催化剂1b上氢键的数目更多,其空间位阻也比二苯基取代的1c的空间位阻更小,手性胍1a能以最高的产率(89%)和ee值(91%)获得螺环己酮氧化吲哚衍生物,因此确定手性胍为该反应的最佳催化剂.在此基础上对催化剂1a的用量进行了筛选,当催化剂1a的物质的量分数为20%时其产率和ee值均达到了最高值.
2.2 反应条件的筛选
以二苯乙烯基甲酮2a、吲哚啉酮3a为底物,以物质的量分数为20%的手性酰基胍1a为催化剂,探讨该反应的最佳反应溶剂、温度和时间(见表2).
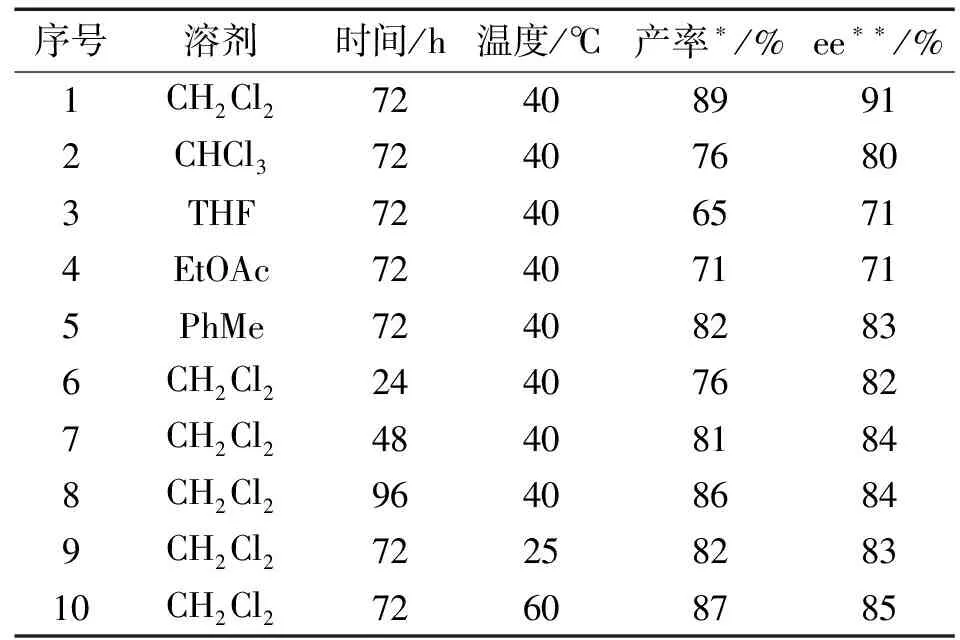
表2 二烯酮2a和吲哚啉酮3a的不对称[5+1]串联的条件筛选研究
首先,在40 ℃和72 h条件下对不同溶剂进行筛选.由表2可知:在甲苯、二氯甲烷、氯仿3种较低极性的溶剂中产物4a都能以中等偏上的产率(76%~89%)和ee值(80%~91%)获得,在二氯甲烷中反应能达到最高的产率(89%)和ee值(91%),而在极性相对较大的乙酸乙酯和四氢呋喃溶剂中反应效果不是很理想,分别仅能得到71%和65%的产率以及71%和71%的ee值.其原因可能是:该反应适合在较低极性的溶剂下反应,且底物和催化剂在二氯甲烷中的溶解性最好.因此选择二氯甲烷为溶剂.然后,对时间和温度进行筛选.在40 ℃下反应72 h,目标产物螺环己烷吲哚啉酮能以89%的产率和91%的ee值获得.故筛选出合成螺环己烷氧化吲哚的最佳反应条件:催化剂为物质的量分数20%的酰基胍1a、溶剂为CH2Cl2、在40 ℃下反应72 h.
2.3 反应底物的扩展
在物质的量分数为20%的催化剂1a、溶剂为CH2Cl2、在40 ℃下反应72 h的最佳反应条件下,对二苯乙烯甲酮和吲哚啉酮的反应底物进行了扩展,共扩展得到15种螺环己酮氧化吲哚衍生物(见表3).
由表3可知:该反应的底物适应性良好,能以75%~91%的产率、73%~93%的ee值和大于20∶1的dr值获得目标产物.在二苯乙烯基甲酮苯环上对位连有吸电子基的底物产率比含有给电子基团的底物产率更高,其原因可能是:二烯酮作为潜在的亲电受体,在苯环上的吸电子基团导致二烯酮的反应活性增加而得到更高的产率.在吲哚啉酮上的基团及位置对反应的产率影响较小,无论是F、Cl还是Br取代,无论是4位取代还是5位取代都能得到中等偏上的产率.二烯酮和吲哚啉酮2者的空间效应和电子性质对目标产物的对映选择性都有一定的影响,空间位阻大的ee值相对较低,但非对映选择性没有太大影响(>20∶1).
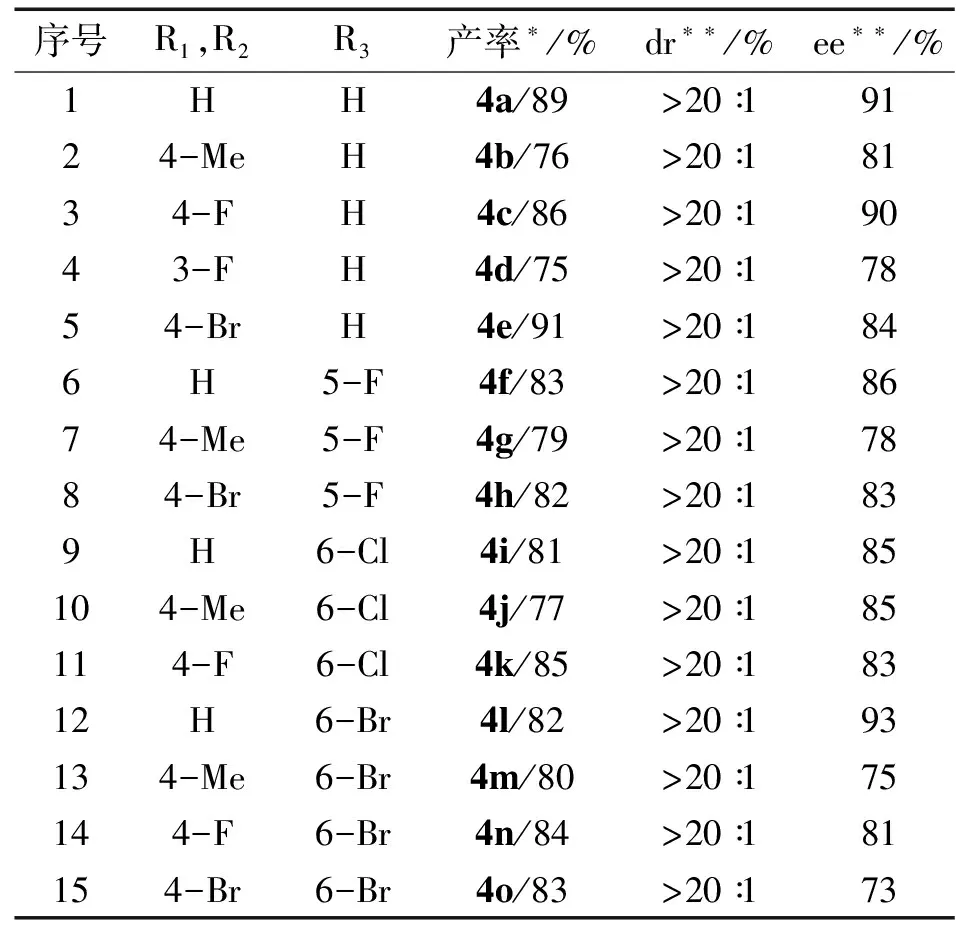
表3 二烯酮2和吲哚啉酮3之间的不对称[5+1]串联
3 结论
本文使用结构上下对称的氢键型催化剂酰基胍催化二苯乙烯基甲酮和氧化吲哚的不对称[5+1]环加成反应,通过1步反应合成了螺环己酮氧化吲哚衍生物,并对反应条件进行了优化.实验结果表明:在以物质的量分数20%的手性胍1a为催化剂、CH2Cl2为溶剂、在40 ℃下反应72 h的条件下,能以91%的产率、93%的ee值获得螺环己酮吲哚啉酮.该反应底物范围广,有较好的普适性,本文扩展合成了15种螺[环己酮-氧化吲哚].该方案为在温和的反应条件下获得具有良好对映体过量值和底物范围的功能化氧化吲哚支架提供了途径.
猜你喜欢
中国典型病例大全(2022年10期)2022-05-10
炼油与化工(2022年1期)2022-03-08
科技研究·理论版(2021年22期)2021-04-18
中国应急管理科学(2021年9期)2021-03-16
教育周报·教育论坛(2020年3期)2020-10-21
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
科技资讯(2017年12期)2017-06-09
