
多羧酸受体化合物的差异性结构构筑与合成线路优化*
2022-10-22 02:38成昭,郑蕾,徐玥,何昊
化学工程师 2022年10期
成 昭,郑 蕾,徐 玥,何 昊
(西安医学院 药学院,陕西 西安 710021)
作为一种被广泛应用的配体,多羧酸受体能够以配位键与许多金属形成配位化合物。典型的多羧酸受体,如乙二胺四乙酸(ethylenediamine tetraacetic acid,EDTA)、二(β-氨基乙基)乙二醇醚-N,N,N',N'-四乙酸(ethylene glycol tetraacetic acid,EGTA)以及1,2-二(2-氨基苯氧基)乙烷-N,N,N',N'-四乙酸(1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid,BAPTA)[1-3]等,应用广泛、被人们所熟知。在多羧酸受体与目标物的识别过程中,受体的多条柔性羧酸链能够形成包合空腔,将目标物包合于其中。包合空腔的尺寸与其对目标物的包合强度,则依赖于受体分子的羧酸链结构。此外,多羧酸受体对目标物的识别与响应作用,还被受体分子的电子云密度所影响[4,5]。如何基于受体与目标物在分子水平的结合机制、进行多羧酸受体的定向结构构筑与合成,仍是该领域的研究难点。
进行识别位点数目与空间排布的差异性设计,定向构筑一系列多羧酸受体T6~T10(图1)。基于电子效应与识别位点设计,调控受体T6~T10所形成的包合空腔尺寸,调节T6~T10与目标物的结合强度[6,7],为阐明多羧酸受体与目标物的可能作用机制提供研究基础。
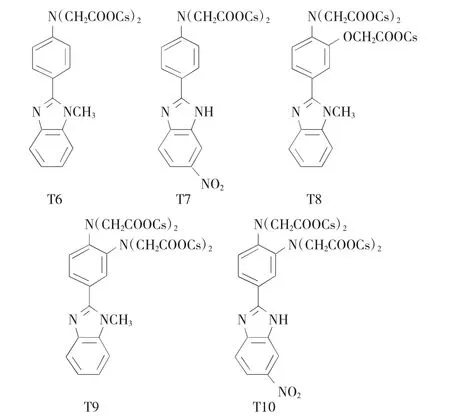
图1 多羧酸受体T6~T10的结构Fig.1 Structures of the polycarboxyl-acid receptors T6~T10
1 实验部分
1.1 仪器与试剂
XT-4型显微熔点仪(北京泰克);TENSOR T-27型傅里叶变换红外光谱仪(美国布鲁克);AVANCE III 400MHz型超导核磁共振波谱仪(美国布鲁克);microTOF-QⅡESI-Q-TOF LC/MS/MS型飞行时间-质联仪(美国布鲁克)。
苯胺、邻氨基酚、邻苯二胺、N-甲基邻苯二胺、4-硝基邻苯二胺、溴乙酸甲酯、二异丙基乙胺、POCl3,均为化学纯,国药集团化学试剂有限公司;N,N-二甲基甲酰胺(DMF)、乙腈、甲醇、无水乙醇、乙酸乙酯、石油醚、无水K2CO3、无水Na2SO4,均为化学纯,天津市天力化学试剂有限公司。
1.2 合成路线
以苯胺、邻氨基酚、邻苯二胺、N-甲基邻苯二胺、4-硝基邻苯二胺、溴乙酸甲酯、POCl3等为原料,经4步合成得到一系列多羧酸受体(化合物T6~T10),合成路线(以T8为例)见图2。
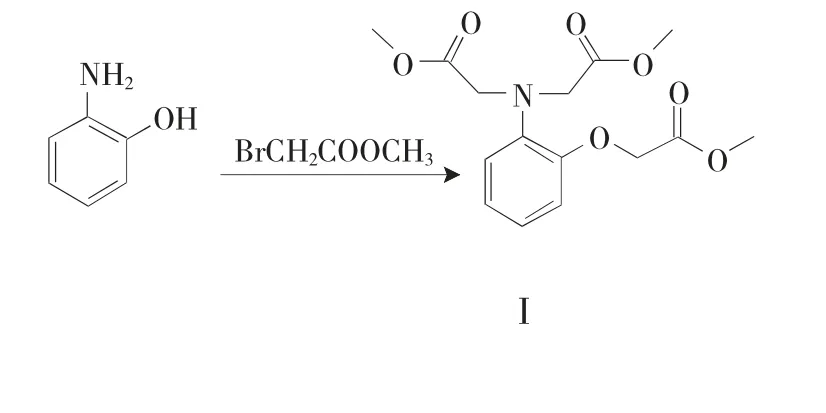
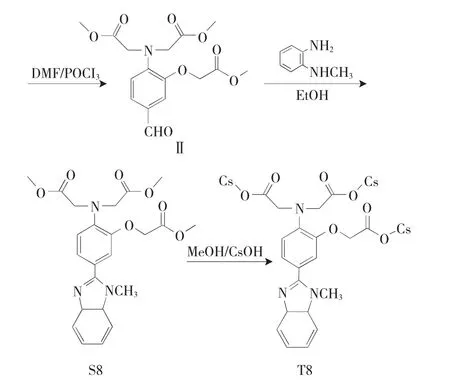
图2 多羧酸受体T8的合成路线Fig.2 Synthetic route of the polycarboxyl-acid receptor T8
1.2.1 化合物I的合成 合成路线中的化合物I包括N,N-二乙酸甲酯基苯胺,1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚,1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯,合成方法如下。
(1)N,N-二乙酸甲酯基苯胺 将苯胺(2.28mL,0.025mol)、二异丙基乙胺(21.75mL,0.125mol)、溴乙酸甲酯(11.75mL,0.125mol)于25 mL乙腈中混合均匀,80℃反应12h。待反应结束、冷却,以100mL冰水稀释、二氯甲烷(100mL×3次)萃取,无水Na2SO4干燥、旋干,柱分离(V乙酸乙酯∶V石油醚=1∶3为洗脱剂),得到N,N-二乙酸甲酯基苯胺。
(2)1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚,1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯 合成方法类似N,N-二乙酸甲酯基-苯胺,反应物投入量分别为2-氨基苯酚(1.09g,0.01mol)、二异丙基乙胺(13.05mL,0.075mol)、溴乙酸甲酯(7.05mL,0.075mol);邻苯二胺(1.08g,0.01mol)、二异丙基乙胺(17.4mL,0.1mol)、溴乙酸甲酯(9.4mL,0.1mol)。
1.2.2 化合物II的合成 合成路线中的化合物II包括4-甲酰基-N,N-二乙酸甲酯基苯胺,5-甲酰基-1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚,5-甲酰基-1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚,合成方法如下。
5~10℃下,将POCl3(2.4mL)缓慢滴加于20mL DMF中,再将上述POCl3/DMF混合物滴加至化合物I(0.01mol)的20mL DMF溶液中,75℃反应4h。待反应结束、冷却,倒入100mL冰水中,抽滤,固体粗品柱分离(V乙酸乙酯∶V石油醚=1∶1为洗脱剂),得到产物II。
化合物I用量N,N-二乙酸甲酯基苯胺(2.37g,0.01mol),1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚(3.25g,0.01mol),1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯(3.96g,0.01mol)。
1.2.3 化合物S6~S10的合成 将化合物II(1mmol)与N-甲基邻苯二胺(0.12g,1mmol,制备S6/S8/S9)或4-硝基邻苯二胺(0.16g,1mmol,制备S7/S10)溶于20mL无水乙醇中,78℃反应4h,待反应结束、冷却,旋干,柱分离(V乙酸乙酯∶V石油醚=2∶1为洗脱剂),得到化合物S6~S10。
化合物II用量4-甲酰基-N,N-二乙酸甲酯基苯胺(0.27g,1mmol),5-甲酰基-1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚(0.35g,1mmol),5-甲酰基-1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚(0.42g,1mmol)。
1.2.4 化合物T6~T10的合成 当应用于水系环境时,乙酸甲酯形式的多羧酸配体S6~S10可水解为其羧酸盐形式,即化合物T6~T10。水解方法为:将S6(0.0367g,0.1mmol)、CsOH·H2O(0.1679g,1mmol)于10mL无水甲醇中混合均匀,80℃反应12h。待反应结束、冷却,旋干,得到多羧酸配体S6的铯盐,即化合物T6。
T7~T10的制备方法类似,反应用量为S7(0.0398g,0.1mmol)、S8(0.0455g,0.1mmol)、S9(0.0526g,0.1mmol)、S10(0.0557g,0.1mmol)。
1.3 结构表征
IR采用KBr压片法对各步合成产物进行IR测试,波数范围4000~400cm-1。
1H NMR以TMS为内标、CDCl3为溶剂,进行400MHz的氢核磁共振分析。经1H NMR、IR与MS表征,S6~S10结构正确,经合成引入的羰基、醚键等官能团均呈现其特征吸收,特征氢位移均能由1H NMR进行归属,符合预期结构设计,各化合物的结构表征数据如下。
(1)N,N-二乙酸甲酯基苯胺 淡黄色油状物,产率88%。1H NMR(CDCl3,400MHz),δ:6.75(t,J=8.0Hz,2H,Ar-H),6.31(t,J=7.3Hz,1H,Ar-H),6.14(d,J=8.4Hz,2H,Ar-H),3.67(s,4H,2×-CH2-),3.28(s,6H,2×-CH3)。IR(KBr),ν,cm-1:3031.33(ν=C-H),2952.76、2849.55(νC-H),1742.01(νC=O),1601.25、1504.75(νC=C),1270.22(νC-O),1204.01(νC-N),752.82、693.38cm-1(δ=C-H)。ESI-MS,m/z:260.0891[M+Na]+。
(2)1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚 白色粉末,产率89%,熔点65~66℃。1H NMR(CDCl3,400MHz),δ:6.92(d,J=3.9Hz,2H,Ar-H),6.81(d,J=7.7Hz,2H,Ar-H),4.67(s,2H,-CH2-),4.22(s,4H,2×-CH2-),3.79(s,3H,-CH3),3.73(s,6H,2×-CH3)。IR(KBr),ν,cm-1:2991.62、2953.79(νC-H),1750.68(νC=O),1590.23、1511.35(νC=C),1202.91(νC-O),750.34(δ=C-H)。ESI-MS,m/z:348.1055[M+Na]+。
(3)1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯 白色粉末,产率89%,熔点135~136℃。1H NMR(CDCl3,400MHz),δ:7.05~7.09(m,2H,Ar-H),6.92~6.98(m,2H,Ar-H),4.32(s,8H,4×-CH2-),3.66(s,12H,4×-CH3)。IR(KBr),ν,cm-1:2954.99(νC-H),1737.74(νC=O),1595.73、1498.15(νC=C),1206.20(νC-O),768.23cm-1(δ=C-H)。ESI-MS,m/z:419.1420[M+Na]+。
(4)4-甲酰基-N,N-二乙酸甲酯基苯胺 淡黄色粉末,产率85%,熔点58~60℃。1H NMR(CDCl3,400MHz),δ:9.79(s,1H,-CHO),7.76(d,J=7.8Hz,2H,Ar-H),6.66(d,J=8.0Hz,2H,Ar-H),4.23(s,4H,2×-CH2-),3.79(s,6H,2×-CH3)。IR(KBr),ν,cm-1:2824.47、2743.11(ν-CHO),1746.67(νC=O),1599.54、1523.10(νC=C),1207.48(νC-O),814.96cm-1(δ=C-H)。ESI-MS,m/z:288.0847[M+Na]+。
(5)5-甲酰基-1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚 淡黄色粉末,产率82%,熔点91~92℃。1H NMR(CDCl3,400MHz),δ:9.78(s,1H,-CHO),7.41(d,J=8.2Hz,1H,Ar-H),6.80(d,J=8.2Hz,2H,Ar-H),4.67(s,2H,-CH2-),4.29(s,4H,2×-CH2-),3.81(s,3H,-CH3),3.79(s,6H,2×-CH3)。IR(KBr),ν,cm-1:3078.89(ν=C-H),1747.87、1681.41(νC=O),1600.55、1519.89(νC=C),1212.84、1168.57(νC-O),869.87、801.79、760.94(δ=C-H)。ESI-MS,m/z:376.1000[M+Na]+。
(6)4-甲酰基-1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯 淡黄色粉末,产率88%,熔点114~115℃。1H NMR(CDCl3,400MHz),δ:9.84(s,1H,-CHO),7.60(s,1H,Ar-H),7.50(d,J=8.2Hz,1H,Ar-H),7.11(d,J=8.6Hz,1H,Ar-H),4.44(s,4H,2×-CH2-),4.30(s,4H,2×-CH2-),3.69(s,6H,2×-CH3),3.67(s,6H,2×-CH3)。IR(KBr),ν,cm-1:3003.45(ν=C-H),2955.46、2919.72(νC-H),1740.53(νC=O),1593.81、1505.48(νC=C),1207.25(νC-O),906.75、834.22cm-1(δ=C-H)。ESI-MS,m/z:447.1378[M+Na]+。
(7)4-(N-甲基苯并咪唑)-N,N-二乙酸甲酯基苯胺(S6)黄色固体,产率70%,熔点170~172℃。1H NMR(400MHz,CDCl3),δ:7.86(d,J=8.0Hz,2H,Ar-H),7.46~7.49(m,2H,Ar-H),7.11~7.14(m,2H,Ar-H),6.61(d,J=7.3Hz,2H,Ar-H),4.14(s,4H,2×-CH2-),3.71(s,6H,2×-CH3),3.26(s,3H,-CH3)。IR ν:3389.23(νN-H),2924.26(νC-H),1735.15(νC=O),1606.24、1509.23、1473.08(νC=C),1479.7cm-1(νN=H),1195.39(νC-O),757.17(δ=C-H)。ESI-MS m/z:368.1610[M+H]+。
(8)4-(4-硝基苯并咪唑)-N,N-二乙酸甲酯基苯胺(S7)红色固体,产率68%,熔点132~134℃。1H NMR(400MHz,CDCl3)δ:8.45(s,1H,Ar-H),8.15(d,J=7.6Hz,1H,Ar-H),7.91(d,J=7.9Hz,2H,Ar-H),7.60(d,J=8.0Hz,1H,Ar-H),6.87(d,J=7.6Hz,2H,Ar-H),5.30(s,1H,-NH-),4.20(s,4H,2×-CH2-),3.79(s,6H,2×-CH3)。IR(KBr),ν,cm-1:3387.06(νN-H),2957.04、2920.92、2851.78(νC-H),1741.76(νC=O),1610.92、1497.82(νC=C),1176.05(νC-O),815.66、737.75(δ=C-H)。ESI-MS,m/z:421.1116[M+Na]+。
(9)5-(N-甲基苯并咪唑)-1-O-2-N,N-三乙酸甲酯基-2-氨基苯酚(S8)黄色固体,产率73%,熔点147~150℃。1H NMR(400MHz,CDCl3),δ:7.58(dd,J=6.0,3.1Hz,4H,Ar-H),6.99(d,J=7.6Hz,2H,Ar-H),6.81(s,1H),4.89(s,2H,-CH2-),4.31(s,4H,2×-CH2-),3.78(s,3H,-CH3),3.73(s,6H,2×-CH3),3.56(s,3H,-CH3)。ESI-MS,m/z:456.1763[M+H]+。
(10)4-(N-甲基苯并咪唑)-1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯(S9)黄色固体,产率67%,熔点128~129℃。1H NMR(400MHz,CDCl3),δ:7.87(s,1H,Ar-H),7.66~7.68(m,1H,Ar-H),7.57~7.58(m,2H,Ar-H),7.24~7.26(m,2H,Ar-H),7.17(d,J=7.6Hz,1H,Ar-H),4.33(s,4H,2×-CH2-),4.31(s,4H,2×-CH2-),3.88(s,6H,2×-CH3),3.87(s,6H,2×-CH3),3.65(s,3H,-CH3)。IR(KBr),ν,cm-1:3171.92(νN-H),3022.32(ν=C-H),2975.51(νC-H),1682.81(νC=O),1019.17(νC-O),751.05(δ=C-H)。ESI-MS,m/z:527.2139[M+H]+。
(11)4-(4-硝基苯并咪唑)-1-N,N-2-N',N'-四乙酸甲酯基-1,2-二氨基苯(S10) 红色固体,产率70%,熔点182~183℃。1H NMR(400MHz,CDCl3),δ:7.66~7.68(m,1H,Ar-H),7.52~7.53(m,2H,Ar-H),7.22~7.24(m,2H,Ar-H),7.15(d,1H,J=8.2Hz,Ar-H),4.57(s,1H,-NH-),4.35(s,4H,2×-CH2-),4.33(s,4H,2×-CH2-),3.88(s,6H,2×-CH3),3.87(s,6H,2×-CH3)。IR(KBr),ν,cm-1:3436.29(νN-H),2959.73、2928.68、2871.10(νC-H),1728.08(νC=O),1594.21(νC=C),1125.12、1073.67(νC-O),743.69(δ=C-H)。ESI-MS,m/z:558.1832[M+H]+。
2 结果与讨论
多羧酸配体的合成路径分为4步,其中,第3步苯并咪唑缩合反应的产物收率相对较低,为提高合成路径总产率,进行了缩合步骤的反应影响因素考察、线路优化与机制分析,得到了较优缩合反应条件。
2.1 溶剂对苯并咪唑缩合反应的影响
控制反应物用量为4-甲酰基-N,N-二乙酸甲酯基苯胺(0.27g,1mmol)与N-甲基邻苯二胺(0.12g,1mmol),以S6的合成为例,进行苯并咪唑缩合反应的反应溶剂考察。在二氯甲烷、乙醚、四氢呋喃等10种溶剂体系中[8,9](表1),分别考察反应4h时以提纯产物质量计算的产物收率,进而实现反应溶剂优化与筛选[10,11]。

表1 溶剂对苯并咪唑缩合反应的影响Tab.1 Influence of solvents on the condensation reaction of benzimidazole
1,4-二氧六环做溶剂时,S6收率可达到90%,无水乙醇作反应溶剂时,收率次之,达到70%。而1,4-二氧六环遇高热或氧化剂均不稳定,光照条件下或接触氧气时,因可能生成过氧化物,1,4-二氧六环还具有潜在爆炸危险。经综合考虑与反复实验验证,最终选取无水乙醇为反应溶剂,应用于乙酸甲酯型配体分子S6~S10的合成时,无水乙醇反应体系的收率均可达到约70%。
2.2 苯并咪唑缩合反应的反应机理
以取代邻苯二胺(N-甲基邻苯二胺、4-硝基邻苯二胺)与化合物II(芳香醛)为反应物,通过缩合反应制备S6~S10。其中,缩合反应的温度考察实验显示,低温冰浴条件下,反应4h的缩合产物为油状物(a),放置数天后,油状物发生明显的性状改变,得到新的黄色固体产物(b)。分别进行上述化合物(a)、(b)的紫外-可见与红外光谱分析,特征谱带均显示出明显差异。紫外-可见光谱中,化合物(a)的吸收谱带峰值分别位于307.0、294.5、248.0以及212.0nm,而(b)的吸收谱带峰值为291.0、245.0和212.0nm。红外光谱中,(a)的特征吸收谱带为3066.8cm-1(ν=C-H)、1681.8cm-1(νN=H)、1030.1cm-1(νC-O-C),而化合物(b)的氮氢伸缩振动为1479.7cm-1(νN=H),较化合物(a)的相应特征峰位置红移202.1cm-1。综合光谱分析结果,推测该反应的可能中间产物(a)为席夫碱类化合物,(b)为缩合反应产物苯并咪唑结构(图3),红外光谱特征氮氢伸缩振动的红移,可能是由于咪唑环上的碳氮键介于碳氮单键与碳氮双键之间,相比碳氮单键有所红移。另据文献报道[12-14],该反应的苯并咪唑缩合产物受反应热力学与动力学因素影响较大[15]。芳香醛与取代邻苯二胺先加成后消去、给出席夫碱产物,继而发生关环、得到苯并咪唑缩合产物。(a)席夫碱产物是速率产物与热力学产物,在反应中先生成,低温条件亦可稳定存在。(b)苯并咪唑缩合产物较(a)席夫碱产物稳定,升高反应温度或延长反应时间即能得到,经反应温度与溶剂选择,最终确定该缩合反应的最优反应条件为无水乙醇反应体系、78℃下进行回流反应,反应时长为4h。
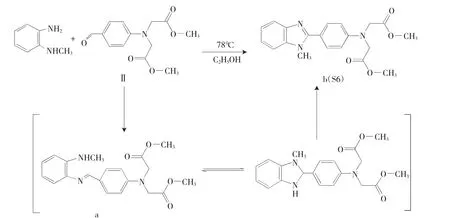
图3 苯并咪唑缩合反应的反应机理Fig.3 Reaction mechanism of the benzimidazole condensation
3 结论
针对多羧酸受体识别位点、包合空腔与其对目标物的包合强度等研究热点与难点,进行多羧酸受体的定向结构构筑与合成。设计空间排布与数目均具有差异性的羧酸柔性链作为多羧酸受体识别位点,以给电子、吸电子基团引入调节多羧酸受体与目标物结合程度的强弱,合成得到多羧酸受体分子T6~T10。此外,针对合成路线中的关键步骤——缩合,进行反应条件优化与反应机制研究,阐明了反应机制,得到了较优的合成反应条件。通过多羧酸受体的差异性结构设计、合成优化、机制研究,为未来工作中进一步阐明多羧酸受体进行目标物识别、结合的可能作用机制提供了研究基础。
猜你喜欢
含能材料(2022年10期)2022-10-22
纺织检测与标准(2021年3期)2021-12-03
中国畜禽种业(2021年9期)2021-09-22
建材发展导向(2021年7期)2021-07-16
建材发展导向(2021年24期)2021-02-12
当代化工(2020年9期)2020-11-30
建材发展导向(2019年5期)2019-09-09
染整技术(2019年2期)2019-04-20
润滑与密封(2019年3期)2019-03-22
分析化学(2017年12期)2017-12-25
