
祛风壮骨颗粒浓缩工艺及质量评价研究 *
2022-11-23 06:08李昕沛吕琴琴刘建峰刘军锋段海洁张红梁文王吉利党秋萍
陕西中医药大学学报 2022年6期
李昕沛 吕琴琴 刘建峰 刘军锋 段海洁 张红 梁文 王吉利 党秋萍
(1.西安市第五医院,陕西 西安 710082;2.陕西省中医药研究院,陕西 西安 710003)
祛风壮骨颗粒具有补肾、强筋骨、止痛的功效,主要用于骨关节炎病、骨质增生、骨质疏松症。祛风壮骨颗粒原剂型为糖浆剂,在临床上应用取得了较满意的疗效,获得了患者的好评,取得了较好的社会效益和经济效益,是我院骨性关节炎临床治疗的重要制剂[1-2]。该方由熟地黄、鹿衔草、鸡血藤、骨碎补、肉苁蓉(炙)、淫羊藿(油炙)、莱菔子(炒)七味药组成,其中淫羊藿的特征性成分为淫羊藿苷,现代药理研究表明淫羊藿苷具有抗衰老、抗肿瘤、补肾壮阳等功效,能增加心脑血管血流量,促进造血功能、增强免疫功能及骨代谢[3]。本研究以淫羊藿苷含量为指标,考察祛风壮骨颗粒大生产中常用的浓缩和干燥方式对淫羊藿苷的影响,为大生产中的工艺优化提供参考。对熟地黄、鸡血藤、骨碎补、淫羊藿(油炙)、莱菔子(炒)进行薄层色谱鉴别,并采用HPLC法对淫羊藿苷进行含量测定,以有效控制本品的内在质量。
1 仪器与试药
1.1仪器 HH-2型数显恒温水浴锅(国华电器有限公司);OSB-2100旋转蒸发器(上海爱朗仪器有限公司);电热鼓风干燥箱(北京科伟永鑫实验仪器设备厂);ZKF040型真空干燥箱(上海实验仪器厂有限公司);Waters2695高效液相色谱仪(美国沃特斯,包括自动进样器,四元泵,柱温箱,2489紫外检测器);GB204电子天平(瑞士梅特勒托利多);SHB-Ⅱ循环水式多用真空泵(郑州长城科工贸有限公司);SB-3200D超声清洗机(宁波新芝生物科技股份有限公司);硅胶G薄层板(烟台江友硅胶开发有限公司);硅胶G(青岛海浪硅胶干燥剂厂,批号:111101);硅胶GF254薄层板(烟台江友硅胶开发有限公司);甲醇、乙腈(Fisher公司,色谱纯)。
1.2试药 淫羊藿苷(中国食品药品检定研究院,批号:110737-200413);柚皮苷(中国药品生物制品检定所,批号:110722-200610);熟地黄对照药材(中国食品药品检定研究院,批号:121196-201105);鸡血藤对照药材(中国药品生物制品检定所,批号:1173-200001);骨碎补对照药材(中国药品生物制品检定所,批号:1169-200001);莱菔子对照药材(中国药品生物制品检定所,批号:120928-201007);祛风壮骨颗粒(医院制剂室自制,批号:1105211、1105212、1105213)。
2 浓缩工艺
2.1复方提取液的制备 按处方比例称取熟地黄15 g,鹿衔草10 g,鸡血藤10 g,骨碎补10 g,肉苁蓉(炙)10 g,淫羊藿(油炙)10 g,莱菔子(炒)10 g,其中莱菔子用布包和其余六味,加水煎煮三次,第一次加8倍量水,煎煮2.5小时;第二、第三次加6倍量水,煎煮1.5小时,合并煎煮液,滤过,得复方提取液备用。
2.2淫羊藿苷含量测定方法[1]
2.2.1对照品溶液配制 精密称取淫羊藿苷对照品1.38 mg,置于10 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀后即得。
2.2.2供试品溶液的制备 精密移取复方提取液2 mL,加60%乙醇稀释并定容至10 mL棕色容量瓶中,摇匀,取上清液过滤即得。
2.2.3色谱条件及系统适应性实验 色谱柱:SunFire C18(4.6 mm×150 mm,5 μm);流动相乙腈-水溶液(30∶70),等度洗脱7.5 min;流速1 mL·min-1;检测波长270 nm;进样量10 μL;柱温20 ℃。
2.2.4线性范围考察 分别精密吸取“2.2.1”项下制备好的对照品溶液1、3、5、7、9 μL,在“2.2.3”项下条件测定峰面积。以对照品质量浓度 (X)与峰面积 (Y)作标准曲线,得对照品淫羊藿苷的回归方程为Y=2×106X+3002.4,r=0.999,在0.138~1.242μg范围内峰面积与对照品质量浓度呈良好线性关系。
2.2.5精密度试验 精密吸取对照品溶液1份,连续重复进样6次,按“2.2.3”项下条件测定峰面积,结果淫羊藿苷峰面积的RSD为1.40%,表明仪器精密度良好。
2.2.6稳定性试验 取“2.2.2”项下供试品溶液,分别于 0、2、4、6、8、10、12 h 按“2.2.3”项下色谱条件测定峰面积,结果淫羊藿苷峰面积的RSD为1.71%,表明本品12 h内稳定性良好。
2.2.7重复性试验 取同一批处方量药材6份,按照“2.2.2”项下方法制备,在“2.2.3”项下条件测定,结果淫羊藿苷峰面积的RSD为1.8%,表明本方法重复性好。
2.2.8加样回收试验 称取处方量药材6份,每份加淫羊藿苷适量,按“2.2.2”项下方法制备,再按“2.2.3”项下条件测定峰面积,计算回收率,淫羊藿苷的平均回收率为98.5%,RSD为1.06%。
2.3浓缩工艺的考察 将复方提取液平均分为3份,第一份不做处理,第二份置于旋转蒸发仪中60℃(-0.07 MPa~-0.08 MPa)条件下浓缩到60 mL,第三份置于100℃水浴条件下浓缩到60 mL,备用。分别取提取液适量,置10 mL容量瓶中,加60%乙醇至刻度,摇匀,得供试品溶液,测定淫羊藿苷的含量,并计算淫羊藿苷的转移率(见表1)。
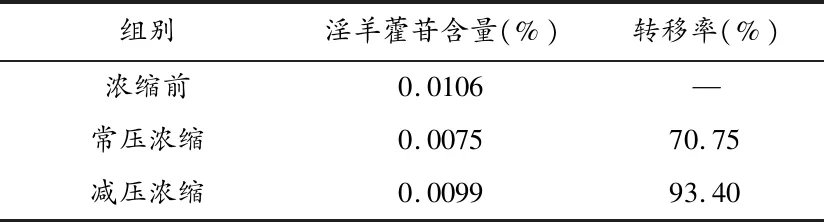
表1 浓缩条件考察结果表
常用的浓缩方法包括常压浓缩和减压浓缩,常压浓缩由于操作时间较长,容易破坏成分,转移率比减压浓缩低,因此选用减压浓缩。
2.4干燥工艺的考察 将上述减压浓缩的稠膏,分成2份,第一份置于80 ℃电热鼓风干燥箱中干燥,第二份置于60 ℃真空干燥箱中干燥,备用。取稠膏适量用60%乙醇溶解后,置10 mL容量瓶中,加60%乙醇至刻度,摇匀,得供试品溶液,测定淫羊藿苷的含量,并计算淫羊藿苷的转移率(见表2)。
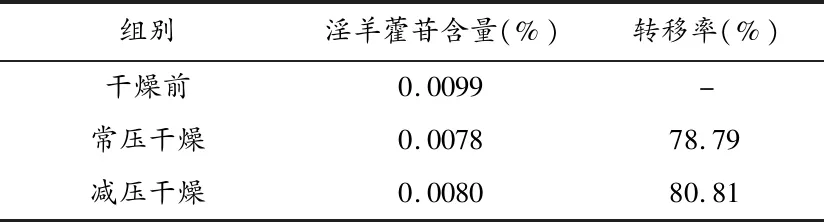
表2 干燥条件考察结果表
实验表明,在60 ℃、-0.08 MPa~-0.09 MPa条件下干燥较佳。
3 质量评价
3.1鉴别
3.1.1熟地黄的薄层鉴别[1,4]取本品5 g,熟地黄对照药材0.5 g和熟地黄阴性样品5 g,加20 mL乙醇超声提取30 min,滤液蒸干后加2 mL甲醇溶解,分别制成供试品溶液、对照药材溶液和阴性对照溶液。参照薄层色谱法(附录Ⅵ B)试验,吸取上述三种溶液各10 μL,分别点于同一硅胶GF254薄层板上,以三氯甲烷-甲醇(10∶1)作为展开剂,展开,取出,晾干后在紫外光灯(254 nm)下检视。供试品色谱中,在与对照药材薄层色谱相应的位置显相同颜色荧光斑点,阴性无干扰(见图1)。

1,2,3-供试品溶液;4-熟地黄对照药材溶液;5-熟地黄阴性对照溶液
3.1.2鸡血藤的薄层鉴别[1,5]取本品5 g,鸡血藤对照药材0.5 g和鸡血藤阴性样品5 g,加水50 mL溶解后用乙酸乙酯萃取3次,每次20 mL,合并萃取液,蒸干后加乙酸乙酯1 mL使溶解,制取供试品溶液、对照药材溶液和阴性对照溶液。参照薄层色谱法(附录Ⅵ B)试验,吸取上述三种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇(20∶1)为展开剂,展开,取出晾干,放入含浓氨水的展开缸中约3 min后取出,置紫外光灯(254 nm)下检视。供试品色谱中,在与对照药材薄层色谱相应位置显相同颜色的荧光斑点,阴性无干扰(见图2)。
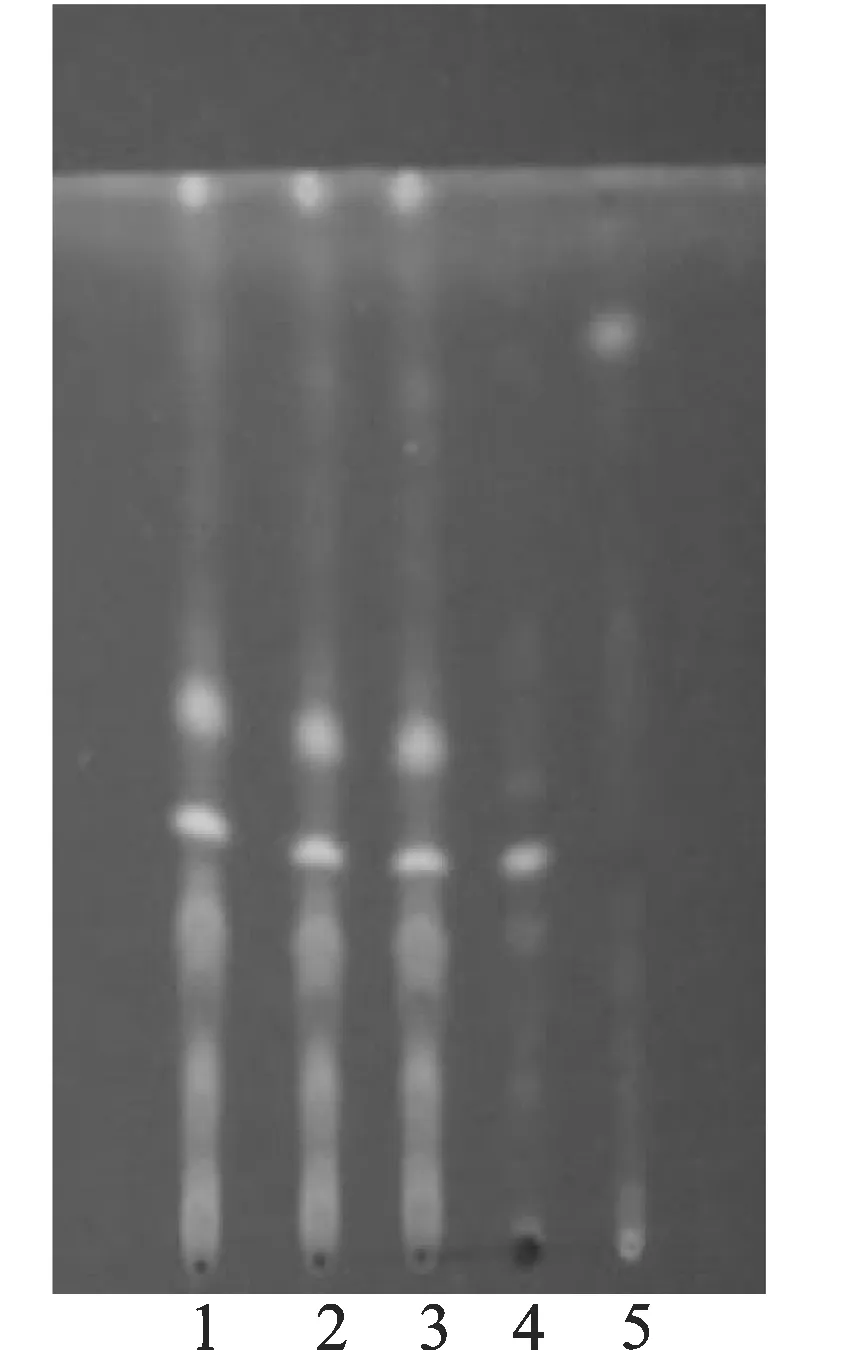
1,2,3-供试品溶液;4-鸡血藤对照药材溶液;5-鸡血藤阴性对照溶液
3.1.3骨碎补的鉴别[1,6]取本品5 g,骨碎补对照药材0.5 g和骨碎补阴性样品5 g,加20 mL乙醇,70 ℃水浴温浸30 min,滤液蒸干,残渣加2 ml乙醇溶解,即得供试品溶液、对照药材溶液和阴性对照溶液。另取柚皮苷对照品0.1 g,加2 mL甲醇溶解,作为对照品溶液;参照薄层色谱法(附录Ⅵ B)试验,吸取上述三种溶液各10 μL,分别点于同一硅胶G薄层板上,以水饱和正丁醇溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材和对照品薄层色谱相应位置上,显相同颜色荧光斑点,阴性无干扰(见图3)。

1,2,3-供试品溶液;4-骨碎补对照药材溶液;5-柚皮苷;6-骨碎补阴性对照溶液
3.1.4淫羊藿的鉴别[1,7-8]称取取本品5 g,淫羊藿阴性样品5 g,加50 mL水溶解后用乙酸乙酯萃取3次,每次20 mL,合并萃取液,蒸干,残渣加1 mL乙酸乙酯溶解,制取供试品溶液、阴性对照溶液。另取淫羊藿苷对照品0.1 g,加2 mL甲醇溶解,即得对照品溶液。参照薄层色谱法(附录ⅥB)试验,吸取上述三种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(6.5∶3.5∶1) 10 ℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷10%硫酸乙醇溶液,于105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照品薄层色谱相应位置显相同颜色荧光斑点,阴性无干扰(见图4)。
3.1.5莱菔子的鉴别[1,9]取本品5 g,对照药材0.5 g和阴性样品5 g,加20 mL乙醇,70 ℃水浴温浸30 min,滤液蒸干,残渣加2 mL乙醇溶解,制取供试品溶液,对照药材溶液和阴性对照溶液。照薄层色谱法(附录Ⅵ B)试验,吸取上述三种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(6.5∶3.5∶1) 10 ℃以下放置的下层溶液为展开剂,展开,取出后晾干,置于有浓氨水的展开缸中约3 min后取出,在紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材薄层色谱相应位置显相同颜色荧光斑点,阴性无干扰(见图5)。

1,2,3-供试品溶液;4-淫羊藿苷对照品溶液;5-淫羊藿阴性对照溶液
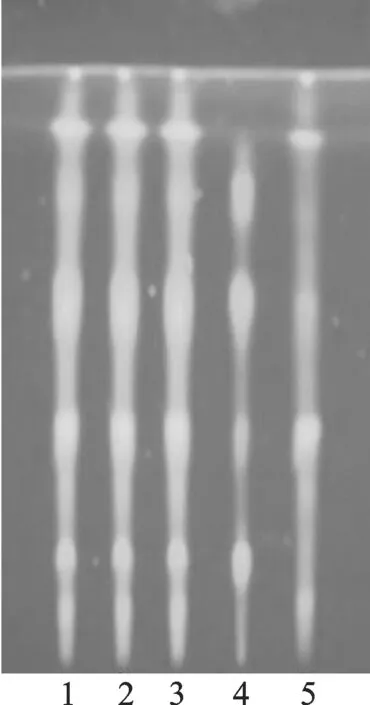
1,2,3-供试品溶液;4-莱菔子对照药材溶液;5-莱菔子阴性对照溶液
3.2含量测定
3.2.1对照品溶液配制 精密称取淫羊藿苷1.38 mg,置10 mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得对照品溶液。
3.2.2供试品溶液制备 取本品约0.5 g,精密称定,置具塞锥形瓶中,精密加入60%乙醇10 mL,密塞,摇匀,称定重量,超声处理40 min,放至室温,再称重,用60%乙醇补足减失的重量,摇匀,滤过即得。
3.2.3色谱条件及系统适应性实验 色谱柱:SunFire C18(4.6 mm×150 mm,5 μm);柱温20 ℃;流动相乙腈-水溶液(30∶70),等度洗脱7.5 min;流速1 mL·min-1;检测波长270 nm;进样量10 μL;对照品溶液和供试品溶液色谱图如图6所示。

图6 对照品及样品的高效液相色谱图
3.2.4线性范围考察 分别精密吸取对照品溶液1、3、5、7和9 μL,按“3.2.3”项下色谱条件进样,测定峰面积。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y),进行线性回归,得淫羊藿苷回归方程Y=2×106X+3002.4,r=0.999,在0.138~1.242 μg范围内峰面积与对照品质量浓度呈良好线性关系。
3.2.5精密度试验 取对照品溶液1份,连续重复进样6次,按“3.2.3”项下色谱条件测定峰面积,测得淫羊藿苷RSD为1.3%,表明该仪器精密度良好。
3.2.6稳定性试验 取“3.2.2”项下供试品溶液,分别于 0、2、4、6、8、10、12 h 进样,按“3.2.3”项下色谱条件测定,结果淫羊藿苷峰面积的RSD为1.2%,表明供试品溶液12 h内稳定性良好。
3.2.7重复性试验 取同一批祛风壮骨颗粒6份,按“3.2.2”项下方法制备供试品溶液,按“3.2.3”项下色谱条件测定,结果淫羊藿苷峰面积的RSD为2.0%,表明该方法重复性良好。
3.2.8加样回收试验 称取同一批祛风壮骨颗粒6份约0.50 g,精密称定,每份加已知量的淫羊藿苷,按“3.2.2”项下方法制备供试品溶液,参照“3.2.3”项下色谱条件分别测定峰面积,计算回收率,结果淫羊藿苷的平均回收率为98.1%,RSD为0.86%,结果见表3。
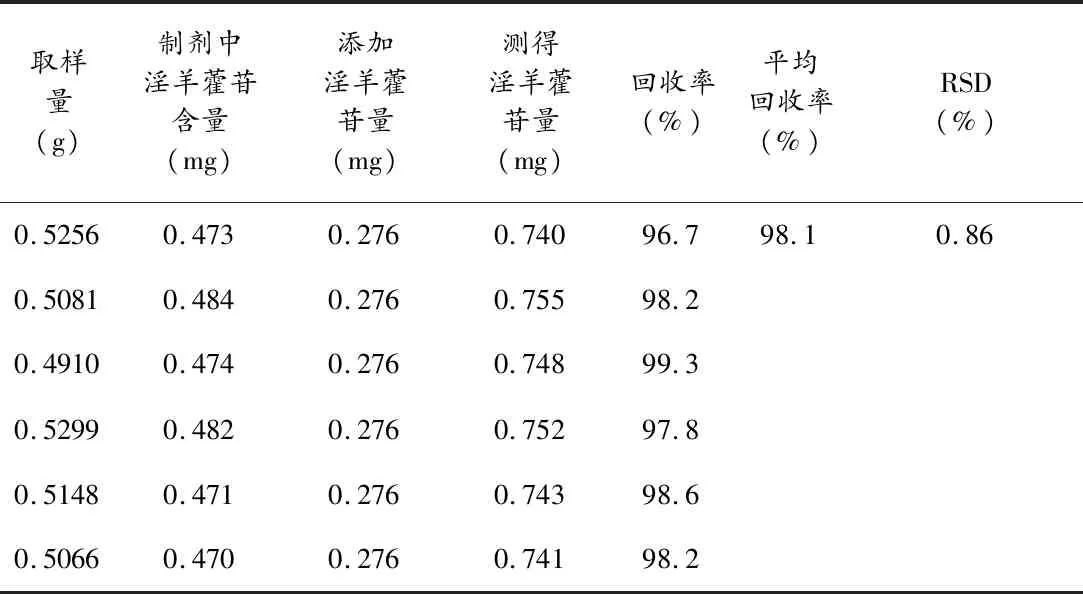
表3 加样回收试验结果
3.2.9含量测定 称取3批祛风壮骨颗粒适量,按“3.2.2”项下方法制备供试品溶液,按“3.2.3”项下方法测定峰面积,计算祛风壮骨颗粒中淫羊藿苷的含量(mg·g-1),结果见表4。
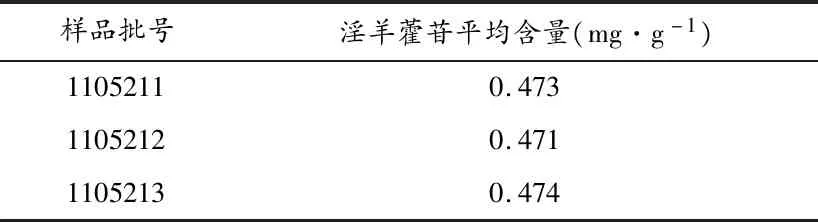
表4 样品含量测定结果
由三批祛风壮骨颗粒含量测定结果可知,本品每批样品含淫羊藿苷较为均匀,表明产品质量稳定。
4 讨论
从表1和表2可见,减压浓缩、干燥时淫羊藿苷转移率高于常压浓缩、干燥,故采用减压浓缩、干燥法进行复方的浓缩及干燥有利于防止活性成分的损失。常压浓缩和常压干燥下,淫羊藿苷的转移率相对较低,可能是因为淫羊藿苷在温度较高的环境中不稳定造成的[10],因此,为了确保活性成分最大限度的被保留,建议在合适温度条件下进行。
建立了5味中药材熟地黄、鸡血藤、骨碎补、淫羊藿(油炙)、莱菔子(炒)的TLC鉴别方法,该法简便,重现性好。采用HPLC法测定了祛风壮骨颗粒中淫羊藿苷的含量,该法准确灵敏、快速简便。本实验建立的薄层鉴别和含量测定方法可用于祛风壮骨颗粒的质量控制[11-14]。
猜你喜欢
药学研究(2021年1期)2021-03-02
世界科学技术-中医药现代化(2021年10期)2021-03-02
中华养生保健(2020年5期)2020-11-16
长寿(2019年9期)2019-10-11
健康前沿(2019年6期)2019-09-10
中成药(2018年12期)2018-12-29
中成药(2018年9期)2018-10-09
中成药(2017年4期)2017-05-17
中国卫生标准管理(2015年4期)2016-01-14
中国中医药现代远程教育(2014年14期)2014-03-01
