
基于CRISPR/Cas9系统Ⅰ型干扰素受体亚单位1基因敲除的Caco-2细胞系的构建
2023-02-20 06:52刘欣奕安妮章青王宏孔翔羽王铭月庞立丽段招军
中国生物制品学杂志 2023年2期
刘欣奕,安妮,章青,王宏,孔翔羽,王铭月,庞立丽,段招军
1.甘肃中医药大学公共卫生学院,甘肃 兰州 730000;2.中国疾病预防控制中心病毒病预防控制所国家卫生健康委员会医学病毒和病毒病重点实验室,北京 102206;3.中国医学科学院医药生物技术研究所,北京 100050
自1957 年从灭活流感病毒感染鸡胚中发现干扰素(interferon,IFN)至今,研究人员相继揭示了IFN的转录诱导机制、作用模式、抗病毒作用等。根据与受体结合方式不同,将IFN 分为Ⅰ、Ⅱ和Ⅲ型。Ⅰ型IFN 包含IFNα、IFNβ、IFNκ、IFNε、IFNω、IFNτ、IFNδ、IFNζ;Ⅱ型IFN包含IFNγ;Ⅲ型IFN包含IFNλ1、IFNλ2、IFNλ3、IFNλ4。在发挥抗病毒作用中,细胞通过胞质途径和跨膜途径,利用模式识别受体识别病原体相关分子模式[1-2],诱导和激活 IFN 产生,之后 IFN 与IFN受体结合,通过信号传导通路引起下游信号的激活[3-4],一部分产生转录因子,通过间接作用,正反馈继续促进IFN的大量产生;另一部分则活化IFN刺激基因(interferon-stimulated genes,ISGs)的表达,产生细胞因子直接发挥抗病毒作用[5-6]。1976 年,人IFN成功治疗了2 例慢性乙型肝炎患者,被美国批准进入相关病毒感染性疾病的临床试验和治疗。目前,IFN 已成为临床针对乙型和丙型肝炎病毒感染的主要治疗制剂[7]。
Ⅰ型 IFN 受体(interferon alpha/beta receptor,IFNAR)由低亲和力IFNAR亚单位1(interferon alpha/beta receptor subunit 1,IFNAR1)和高亲和力IFNAR亚单位2(interferon alpha/beta receptor subunit 2,IFNAR2)基因两个亚基组成[8-9],与IFN 结合后,活化的二者分别继续招募磷酸化非受体蛋白酪氨酸激酶2(nonreceptor protein tyrosine kinase 2,TYK2)和酪氨酸激酶1(janus tyrosine kinase 1,JAK1),从而磷酸化信号传导与转录激活子(signal transducers and activators of transcription,STAT),解离成二聚体,与干扰素调节因子 9(interferon regulatory factor 9,IRF9)形成干扰素刺激转录因子3(interferon-stimulated transcription factor 3,ISGF3)复合物,ISGF3 释放入核信号与核内IFN 刺激反应元件(interferon-sensitive response elements,ISREs)结合,ISG进行转录翻译,最终抑制病原体入侵细胞[10]。其中,IFNAR2作为高亲和力亚基主要作用是与下游JAK1 结合,与IFN 配体形成复合物诱导下游信号传导[11],一旦IFNAR1水平过低,IFNAR2 形成的复合物则不会继续进行下游信号的传导。因此,敲除IFNAR1对于细胞IFN 在天然免疫阶段发挥抗病毒作用机制的研究具有重要意义。
人结肠腺癌细胞Caco-2通过普通培养就可自发分化为单层肠上皮细胞,因此可以很好地模拟人体内小肠上皮细胞环境。由于其能构建与人体极为相似的肠道环境,目前常用于胃肠道感染病毒及相关口服药物早期作用机制研究。本研究利用成簇规律间隔短回文重复序列(clustered regularly interspaced short palinmic repeats,CRISPR)/CRISPR 相关蛋白9(CRISPR-associated protein 9,Cas9)基因编辑技术,构建IFNAR1基因敲除的Caco-2细胞系,并进行外源性IFNβ 刺激试验验证,为探索细胞IFN 受体在抵抗病毒入侵的先天免疫过程中发挥的作用机制提供基因敲除细胞模型。
1 材料与方法
1.1 细胞、菌株及质粒 人结肠腺癌细胞Caco-2、人胚胎肾细胞HEK293T、LentiCRISPRv2质粒由中国疾病预防控制中心病毒病预防控制所病毒性腹泻室保存;感受态大肠埃希菌DH5α 购自擎科生物科技有限公司;质粒psPAX2、VSV-G 由中国医学科学院医药生物技术研究所岑山研究员惠赠。
1.2 主要试剂及仪器 限制性内切酶BsmBⅠ、T4 DNA连接酶购自美国NEB 公司;无内毒素质粒提取试剂盒、基因组DNA 提取试剂盒购自美国OMEGA 公司;胶回收试剂盒购自德国QIAGEN 公司;细胞转染试剂 Thermo LipofectamineTM3000、ABI QuantStudio 5 荧光定量PCR 仪购自美国Thermo 公司;IMDM 培养基、DMEM 培养基、胎牛血清(FBS)、胰酶、嘌呤霉素(Puromycin)购自美国 GIBCO 公司;聚凝胺(Polybrene)购自北京 Solarbio 公司;IFNβ、兔抗 IFNAR1 单克隆抗体、辣根过氧化物酶偶联的鼠抗β-actin 单克隆抗体购自美国abcam 公司;辣根过氧化物酶偶联的山羊抗兔IgG 购自碧云天科技有限公司;2 × Taq PCR StarMix(Dye)购自北京康润诚业生物科技有限公司。
1.3 特异性sgRNA 设计 根据CRISPR/Cas9 技术,通过(https://zlab.bio/guide-design-resources)网站在IFNAR1基因第6个和7个外显子区分别设计sgRNA(single guide RNA),命名为IFNAR1-sgRNA-1和IFNAR1-sgRNA-2,根据LentiCRISPRv2载体BsmBⅠ酶切位点,将sgRNA正义链5′端添加CACC,反义链5′端添加AAAC,使其形成互补的黏性末端。根据sgRNA插入载体位置设计sgRNA插入鉴定引物,根据sgRNA靶向序列位置设计PCR鉴定引物,引物序列见表1。引物由北京擎科生物科技有限公司公司合成。

表1 引物序列Tab.1 Primer sequences
1.4 LentiCRISPRv2-IFNAR1-sgRNA 质粒的构建将LentiCRISPRv2 利用BsmBⅠ酶切,1%琼脂糖凝胶电泳鉴定同时回收酶切后线性化质粒。将设计的sgRNAoligo 利用 ABI QuantStudio 5 荧光定量 PCR 仪将温度从95 ℃降至4 ℃,按照1 ℃/min 的速度进行退火处理;将线性化的LentiCRISPRv2质粒与退火形成的双链sgRNA 利用T4 DNA 连接酶16 ℃连接过夜;连接产物转化大肠埃希菌DH5α 感受态细胞,涂布氨苄抗性(100 mg/mL)琼脂平板,37 ℃孵育过夜;挑取单克隆,送北京擎科生物科技有限公司测序,比对无误后进行去内毒素质粒提取,提取的质粒命名为Lenti-CRISPRv2-IFNAR1-sgRNA-1(简称S1)和LentiCRI-SPRv2-IFNAR1-sgRNA-2(简称S2)。
1.5 慢病毒包装及感染 将HEK293T 细胞按照6 ×105个/孔接种6孔板,长满80%左右时,将S1、S2分别进行慢病毒包装,将S1/S2∶psPAX2∶VSV-G 按照4∶3∶1的比例,利用LipofectamineTM3000转染至HEK293T 细胞中,6 h 后更换为含 20% FBS 的 IMDM培养基,48 h后收集上清,经0.22 μm滤膜过滤备用。将 Caco-2 细胞按 3 × 105个/孔接种 6 孔板,长满30%~50%时,将慢病毒液与含20%FBS的IMDM 培养基按照1∶1比例加至6孔板中,同时加入8 μg/mL polybrene,感染48 h。
1.6 单克隆细胞株筛选及培养 将感染后细胞的培养基更换为含20% FBS 的IMDM,加入终浓度为2 ~3 μg/mL 的嘌呤霉素,进行传代及筛选,周期为7 ~10 d,筛选2 ~ 3 次;待对照组[野生型 Caco-2 细胞(Caco-2-WT)]全部死亡后,将筛选出细胞的培养基更换为含20% FBS 的IMDM 继续挑取单克隆,利用有限稀释法,将细胞消化为单个细胞,按每孔最多1个细胞的密度接种至96 孔板中,37 ℃温箱培养约1周;消化细胞,按2 × 105个/cm2的接种密度移至6孔板中继续传代培养,获得稳定传代的单克隆细胞。
1.7 单克隆细胞株IFNAR1基因鉴定 采用PCR法。提取单克隆细胞株基因组DNA,PCR扩增目的基因片段。PCR反应条件:94 ℃ 2 min;94 ℃ 30 s,55 ℃ 30 s,72 ℃60 s,共35个循环;72 ℃ 5 min。PCR产物经1%琼脂糖凝胶电泳鉴定后,送北京擎科生物科技有限公司测序。
1.8 IFNAR1 蛋白表达的检测 采用Western blot法。冰上裂解Caco-2-IFNAR1-KO1、Caco-2-IFNAR1-KO2 和 Caco-2-WT,提取蛋白,经 12% SDS-PAGE 分离后,转膜仪转至PVDF 膜上,10%脱脂奶粉室温封闭1 h;加入兔抗IFNAR1单克隆抗体(1∶2 000稀释)和辣根过氧化物酶偶联的鼠抗β-actin 单克隆抗体(1∶4 000 稀释),4 ℃孵育过夜;1 × PBST 洗膜 3 次,每次5 min,加入辣根过氧化物酶偶联的山羊抗兔IgG(1∶1 000 稀释),室温孵育1 h;1 × PBST 洗膜3次,每次5 min,化学发光法显影。
1.9 IFN 受体功能检测 采用IFNβ 刺激试验。将Caco-2-IFNAR1-KO1、Caco-2-IFNAR1-KO2和Caco-2-WT细胞株接种至24孔板中,按照0、50 ng/mL的浓度加入IFNβ,试验重复3 次,37 ℃作用6 h。提取细胞总RNA(去除细胞内DNA),反转录获得cDNA,以其为模板进行Real-time PCR,检测CXC 趋化因子配体 10(CXC chemokine ligand 10,CXCL10)和 IFN 刺激基因 20(interferon-stimulated gene 20,ISG20)mRNA水平。反应条件:50 ℃ 2 min;95 ℃ 2 min,95 ℃ 15 s,60 ℃ 1 min,共 40 个循环;95 ℃ 15 s,60 ℃ 1 min,95 ℃ 15 s。以GAPDH作为内参,以野生型 Caco-2 细胞0 ng/mL IFN 刺激组其中1 孔作为对照,采取相对定量法(2-△△Ct)进行定量。
1.10 统计学分析 采用GraphPad Prism 软件进行统计学处理,t检验进行数据差异分析,以P<0.05 为差异有统计学意义。
2 结果
2.1 重组质粒 LentiCRISPRv2-IFNAR1-sgRNA 的鉴定 测序结果显示插入位置均位于BsmBⅠ酶切黏性末端,见图1。
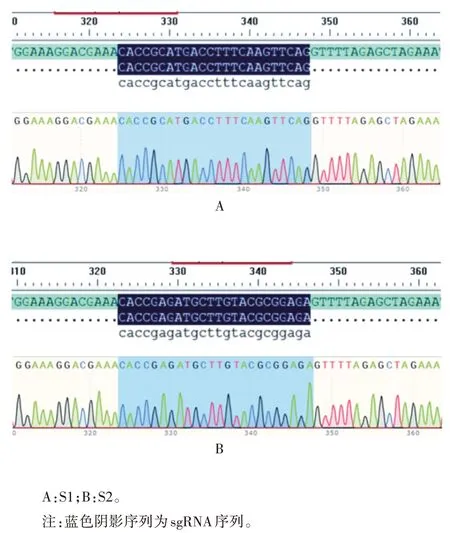
图1 LentiCRISPRv2-IFNAR1-sgRNA质粒测序结果Fig.1 Sequencing result of LentiCRISPRv2-IFNAR1-sgRNA plasmids
2.2IFNAR1敲除Caco-2 细胞株的筛选 最终获得2 株单克隆细胞株:Caco-2-IFNAR1-KO1 和 Caco-2-IFNAR1-KO2。测序结果显示,Caco-2-IFNAR1-KO1IFNAR1第6个外显子发生5 bp缺失,Caco-2-IFNAR1-KO2 第 7 个外显子发生 18 bp 缺失同时有 1 bp 增加,见图2。

图2 IFNAR1敲除Caco-2细胞株的测序结果Fig.2 Sequencing result of IFNAR1 knockout Caco-2 cell lines
2.3IFNAR1敲除 Caco-2 细胞株 IFNAR1 蛋白的表达水平 Western blot 分析显示,Caco-2-WT 细胞中IFNAR1蛋白正常表达,Caco-2-IFNAR1-KO1和Caco-2-IFNAR1-KO2细胞中IFNAR1蛋白未见表达,见图3。表明IFNAR1基因的缺失和插入突变导致IFNAR1阅读框移码,使Caco-2-IFNAR1-KO1和Caco-2-IFNAR1-KO2 细胞不能正常表达IFNAR1 蛋白。成功构建2株IFNAR1 蛋白表达缺失的IFNAR1敲除Caco-2 细胞株。

图3 IFNAR1敲除Caco-2细胞株的Western blot鉴定Fig.3 Identification of IFNAR1 knockout Caco-2 cell lines by Western blot
2.4IFNAR1敲除对IFN 受体功能的影响 结果显示,相比于0 ng/mL IFNβ,在50 ng/mL 外源IFNβ刺激下,Caco-2-IFNAR1-KO1和Caco-2-IFNAR1-KO2细胞CXCL10基因 mRNA 水平(t分别为 0.566 和1.268,P> 0.05)和ISG20基因 mRNA 水平(t分别为1.522 和1.733,P> 0.05)均无明显升高;与Caco-2-WT 细胞相比,在50 ng/mL 外源 IFNβ 刺激下,Caco-2-IFNAR1-KO1 和 Caco-2-IFNAR1-KO2 细胞CXCL10基因mRNA 水平(t分别为6.763 和6.777,P< 0.05)和ISG20基因 mRNA 水平(t分别为 5.664 和 5.653,P<0.05)均显著降低。见图4。表明IFNAR1基因敲除明显抑制了IFNAR下游信号通路的激活。

图4 野生型Caco-2细胞株和IFNAR1敲除Caco-2细胞株中ISG20(A)和CXCL10基因(B)mRNA水平Fig.4 Levels of mRNA of ISG20 gene(A)and CXCL10 gene(B)in wild-type Caco-2 cell line and IFNAR1 knockout Caco-2 cell lines
3 讨论
高效的基因编辑从本质上讲是有针对性的将目标染色体上的DNA 双链进行断裂,诱导DNA 损伤修复。目前在基因编辑技术领域,CRISPR-Cas 作为第三代基因编辑系统[12],无论在便捷性、效率还是成本方面均体现出独有的优势。CRISPR/Cas9 系统是目前最常用的基因编辑工具,将crRNA 和tracrRNA融合在 1条 sgRNA 上,利用sgRNA 识别靶标 DNA[13],极大地提高了操作的针对性和便捷性。本研究利用CRISPR/Cas9 技术敲除 Caco-2 细胞IFNAR1基因,在基因序列和蛋白表达层面验证IFNAR1基因表达被成功敲除,体现了该项技术的可靠性和稳定性。
有研究证实,IFNAR1 作为IFNAR 重要亚基,与IFNAR 结合进一步激活下游JAK-STAT信号通路,对下游信号传导和ISGs 转录激活阶段具有重要作用[14]。IFNAR1 主要通过独立与 IFN 结合,同时招募另一个亚基形成三元复合物才能被激活。在IFNAR1和 IFNAR2 功能试验中发现,IFNAR 中 IFNAR1 数量过低,则不能完全激活下游信号通路[15]。因此,敲除IFNAR1对抑制细胞发挥Ⅰ型IFN 抗病毒作用,具有重要意义。
CXCL10和ISG20作为IFN激活因子,对IFN发挥抗病毒过程起到直接和间接的重要作用。CXCL10又称IFN 诱导蛋白10,是CXC 趋化因子家族的一种细胞因子[16]。作为调节机体炎症和免疫反应的重要趋化因子配体,作用机制为通过与CXCR3受体结合,诱导细胞趋化、细胞凋亡、细胞生长和血管稳定[17]。有文献表明,在感染病毒的个体体液中观察到CXCL10的异常水平,表明CXCL10在细胞应对病毒感染等情况时,自身分泌一种及时应对感染创伤的重要机制[18-20]。IFN作为广谱、非特异性的、高效的抗病毒蛋白,其真正发挥抗病毒作用是通过激发ISGs 实现的[21],细胞产生的IFN 通过旁分泌或自分泌形式,作用到自身或临近细胞,通过与相应受体结合,达到大范围高效率激发下游JAK-STAT 信号通路,诱导多种ISGs 表达[22],从而真正发挥直接抗病毒作用。Ⅰ、Ⅱ、Ⅲ型IFN均可诱导产生ISG20,最初报道在感染乙型肝炎病毒的黑猩猩肝脏内进行病毒清除过程中检测到ISG20[23],表明ISG20 可能对细胞应答病毒感染过程产生重要影响。关于ISG20 对病毒抑制作用研究中,通过过表达该因子,大部分单股正链和负链RNA 病毒均明显被抑制[24]。同时也有研究表明,ISG20能抑制HIV-1和乙型肝炎病毒的复制[25]。本研究也显示,IFNAR1敲除Caco-2 细胞的CXCL10和ISG20的转录激活受到了抑制,提示Caco-2细胞敲除IFNAR1基因后降低了抗病毒能力,为后期提高病毒分离效率及病毒感染滴度提供了途径。
病毒性传染病严重威胁人类健康,是引起人类死亡的主要原因之一。许多肠道感染病毒不易在传代细胞上分离培养,无法大量扩增或病毒滴度难以提高,限制了对病毒株的致病机制的研究,阻碍了病毒疫苗的研制和药物筛选。利用CRISPR/Cas9 技术以及针对IFN 发挥作用的关键通路相关基因的敲除构建稳定表达细胞株,对研究IFN 的抗病毒机制及针对难培养病毒的分离和扩增具有重要意义。虽然该技术对于目前存在的脱靶效应仍然无特别有效的方法,但仍为细胞基因层面的编辑和探索提供了更加便捷、广阔的平台和空间。IFN 受体作为IFN 发挥直接抗病毒作用的重要靶点,一些机制尚不明确,本文通过CRISPR/Cas9基因编辑技术构建了IFNAR1基因敲除Caco-2 细胞系,为研究IFN 通路抗病毒的效果和机制提供了工具,为后续针对其他通路和因子的研究也具有借鉴意义。
猜你喜欢
肝博士(2022年3期)2022-06-30
肝博士(2021年1期)2021-03-29
中国现代医药杂志(2020年10期)2020-12-14
肝博士(2020年4期)2020-09-24
疯狂英语·初中天地(2020年3期)2020-05-21
现代检验医学杂志(2016年3期)2016-11-15
医学研究杂志(2015年11期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
中国当代医药(2015年16期)2015-03-01
