
靶向切割猪Y 染色体的CRISPR/Cas9 载体构建及功能验证
2023-02-22 19:19霍梦飞孟繁明王塑天李颢杨化强
华南农业大学学报 2023年2期
关键词:染色体
霍梦飞 孟繁明 王塑天 李颢 杨化强

摘要: 【目的】通过CRISPR/Cas9 系统对Y 染色体多位点切割,实现目标染色体的敲除,为畜禽性别控制提供新的手段。【方法】以CRISPR/Cas9 技术为基础,寻找Y 染色体上能被sgRNA 特异性识别的多拷贝重复序列,并通过体外切割、定量分析、核型鉴定验证其对靶点的有效性。【结果】所设计的sgRNA 能够在体外实现对靶片段的明显切割,且切割效率均达到了50% 以上。基因的定量分析结果证明了其在细胞水平切割的有效性,且簇状重复序列切割效果明显优于散在重复序列;核型鉴定结果证实了细胞水平猪Y 染色体的丢失。【结论】研究结果为后续构建染色体敲除猪,实现猪的性别控制奠定了基础。
关键词: 猪;CRISPR/Cas9;Y 染色体;性别控制;基因编辑
中图分类号: S813.3文献标志码: A 文章编号: 1001-411X(2023)02-0187-10
染色体作为生物遗传物质的载体,对当前生命遗传研究开展尤为重要。染色体消除技术(Chromosomeelimination technology) 近年来随着基因编辑技术的发展不断进步,在动物模型构建、家畜育种改良中的性别控制、特定染色体的功能研究以及人类非整倍体疾病治疗等领域具有重要的应用价值。目前通过该技术已成功在人、小鼠、斑马鱼等物种上实现了特定染色体的消除[1-3],然而打靶效率低、非靶点的額外失活及操作过程的繁琐性大大制约了该技术的发展[3-5]。但随着新一代CRISPR/Cas9 技术的出现,灵活的靶点选择、较高的编辑效率以及对于靶点多重切割的可能性使该技术的有效运用成为可能。
目前,研究者们已通过锌指核酸酶(Zinc fingernuclease,ZFN) 技术、Cre/loxP 系统、TKNEO 基因敲入等多种方法实现了目标染色体的敲除[5-7]。ZFN 技术通过人工改造的核酸内切酶(锌指蛋白单元) 与特定的DNA 序列结合赋予其精确的打靶功能[8],但其脱靶效应、毒性及锌指结构的低适用性造成其基因操作效率低下[ 9 ];Cre/loxP 系统主要由Cre 重组酶与2 个loxP 位点组成,由于loxP 的位点不同会产生DNA 链的插入、删除、易位等不同重组方式[10],但该系统的使用需要引入特定表达元件且本身会对细胞产生毒性[4, 11];TKNEO 是胸苷激酶(Thymidine kinase,TK) 和新霉素抗性(Neomycinresistance,NEO) 的融合编码基因,利用传统基因打靶技术敲入目标染色体进行药筛可产生自发性染色体缺失[3],但其操作的繁琐性和不确定性使其并不具有广泛的适用性。
CRISPR/Cas9 技术近年来已经成为了基因编辑的主要手段,其由大肠埃希菌Ⅱ型CRISPR/Cas 系统改造而来,由于其引导RNA(Small guide RNA,sgRNA) 序列的灵活性而能对多个位点进行灵活编辑,从而实现基因的定点敲入、敲除或其他多种修饰[12-16]。该系统主要由2 个部分组成,Cas9 蛋白和sgRNA,其中后者负责识别目标靶序列,前者依靠其自身含有的2 个切割域完成对目标序列的切割,之后依靠细胞本身的2 种修复机制(同源重组修复和非同源末端连接) 实现目标序列的精确编辑[17-21]。最新研究表明,CRISPR/Cas9 系统可通过产生多重DNA 双链断裂(Double-strand breakage,DSB) 引起Y 染色体降解[22-23],对这种方法,虽然部分细胞会因自身修复机制保持基因组的稳定性,但是仍有相当的几率可产生特定染色体消除的细胞,通过筛选出此种染色体消除的细胞系作为供体应用于动物克隆,可制备出特定染色体消除的动物,因此达到控制动物本身及其后代的性别进行偏移的目的。
本研究拟通过CRISPR/Cas9 技术,在猪Y 染色体内寻找能够被特异sgRNA 识别的重复序列,进行多位点切割以实现目标染色体的降解。本研究旨在为特定染色体及其携带基因功能的研究提供一种新的工具,同时也为实现大型家畜性别控制提供一种新的技术方案。
1 材料与方法
1.1 材料
本试验所用的CRISPR/Cas9 载体HP180_p x 3 3 0 _ G F P 及猪胎儿成纤维细胞( P o r c i n eembryonic fibroblast,PEF) 由国家生猪种业工程技术研究中心保存。设计的sgRNA 及相关引物由华大基因合成。质粒与基因组DNA 抽提试剂盒、限制性内切酶B p i I 、g R N A 体外转录试剂盒GeneArtTM Precision gRNA Synthesis Kit、荧光定量试剂盒PowerUpTMSYBRTMGreen Master Mix 购自赛默飞公司;T4 DNA 连接酶购自TaKaRa 公司;Cas9 内切酶购自NEB 公司;感受态细胞Trans5α、6×DNA Loading buffer、RNA loading buffer 购自全式金生物。高糖DMEM 培养液、胰酶、胎牛血清均购自Gibco 公司。
1.2 方法
1.2.1 寻找Y 染色体特异靶向重复序列及sgRNA设计 在Ensembl 获取整个Y 染色体注释文件,在Linux 下用awk 命令提取注释文件中的所有基因,并在Ensembl 上将每个基因与全基因组序列进行逐一比对,最终筛选出符合条件的多拷贝基因。利用在线网站CCTop - CRISPR/Cas9 targetonline predictor(https://cctop.cos.uni-heidelberg.de/)对所选择靶基因进行sgRNA 设计,确定最终序列与位点,并送至华大基因合成。
1 . 2 . 2 s g R NA 体外切割验证 对设计好的sgRNA 体外转录进行切割,初步筛选出效率较高的进行载体构建。首先进行靶片段的PCR 扩增。根据本试验靶基因的选择,使用Primer Premier 6 进行相应靶点引物设计,合成目标靶片段。然后根据赛默飞公司的GeneArtTM Precision gRNA SynthesisKit 说明书设计sgRNA 体外转录模板引物,并最终进行G e n e _ I D 为E N S S S C G 0 0 0 0 0 0 4 3 0 5 8 、ENSSSCG00000046447 的转录模板合成(以下简称为43058、46447)。其合成原理(图1) 及相关引物(表1、表2) 如下所示。
体外Cas9 切割验证主要包含3 个过程:Cas9-sgRNA 的自发组装、Cas9-sgRNA 复合体与靶片段的特异结合、Cas9-sgRNA 对靶点进行特异切割。采用2 0 μ L 反应体系: 体外转录s g R NA 模板100 ng,靶片段250 ng,Cas9 蛋白1 μL,10×Cas9Reaction buffer 2 μL,最后加Nuclease-free water 补足至20 μL。
在进行Cas9 体外切割时,反应体系首先加入除靶片段外其他成分进行Cas9 与sgRNA 组装(组装时间30 min),再加入相应靶片段孵育1 h,切割完毕后在反应体系中加入2 μL 的蛋白酶K 再次孵育20 min。最终切割产物加入6×DNA LoadingBuffer 进行20 g/L 的凝胶电泳,分析电泳条带灰度计算切割效率。
1.2.3 CRISPR/Cas9 载体构建 确定有效的sgRNA 后进行真核细胞表达载体构建。首先使用限制性内切酶BpiI 对载体HP180_px330_GFP(图2) 双酶切使其线性化,合成的sgRNA 经退火形成双链由T4 连接酶在16 ℃ 条件下连接1 h,然后热应激转入Trans5α 感受态细胞,于220 r/min、37 ℃培养1 h 后涂布含氨苄青霉素的LB 平板,隔夜培养后挑取单克隆,进行菌液PCR 并送测序,测序正确菌液进行质粒抽提,用于PEF 电转。
1.2.4 PEF 培养与转染 试验所用细胞经鉴定为公猪细胞(使用引物F:TCATAGCTCAAACGATGGACGTG,R:ACACAATGAAAGCGTTCATGGGTC,扩增片段大小为92 bp)。PEF 复苏后加入含体积分数为12% 胎牛血清的高糖DMEM 培养液,置于38 ℃、CO2 体积分数为5% 的培养箱中培养。当复苏后的细胞融合度达到80%~90% 时,用质量分数为0.05% 的胰酶消化后进行细胞计数。取1×106 mL?1 细胞悬液加入1.5 mL离心管,加入100 μL 电转液及目的质粒以Lonza核转仪进行电转,电转程序A003。转染24 h 后,荧光显微镜下拍照并观察转染细胞荧光信号强弱,判断转染效果。
1.2.5 流式分选及单细胞克隆团鉴定 细胞转染2~3 d 后进行流式分选(Fluorescence activating cellsorter,FACS)。细胞分选前经质量分数为0.05% 的胰酶消化,加入含体积分数为2% 胎牛血清的DPBS 重悬,并在上机前经40 μm 细胞筛过滤防止细胞团块的干扰。根据GFP 标记激发波长488 nm分选出阳性细胞(5×104~10×104 个) 并进行极限稀释(梯度稀释降低细胞密度) 培养,细胞培养形成单个细胞团后取部分细胞抽提DNA 进行Y 染色体性别决定区(Sex determination region of Y chromosome,SRY) 基因鉴定。引物信息如表3 所示。
1.2.6 实时定量荧光PCR 检测Y 染色体敲除效率 分选后待细胞融合度达到80%~90% 提取基因组DNA,并设计相应靶基因及SRY 定量引物(表3)观察其DNA 含量是否显著降低。以基因组为模版检测目的基因在DNA 水平的含量是否变化,并以基因组水平的β-actin 为内参。反应体系与反应产物均按照相应试剂盒(PrimeScriptTM RT reagent Kitwith gDNA Eraser 与PowerUpTM SYBRTM GreenMaster Mix) 说明进行。
1.2.7 核型鉴定 对SRY 鑒定为阴性细胞继续扩繁培养,并传代至T25 培养瓶,待细胞生长至融合度为70%~80% 时进行核型分析,判断染色体核型是否缺失。核型分析由杭州卡优太谱生物科技公司进行。
2 结果与分析
2.1 多拷贝基因查询及sgRNA 设计结果
2.1.1 多拷贝基因查询结果 通过Ensembl 的blast 筛选出了35 个位于Y 染色体上的多拷贝基因,统计结果如表4 所示。
通过再次与NCBI 数据库进行比对,以及基于靶点基因组测序的真实结果与sgRNA 设计的实际情况,我们最终选择了网站预测评分较高、符合测序结果且在Y 染色体上重复拷贝数靠前(多位点DSB) 的Gene_ID 为ENSSSCG00000043058、ENSSSCG00000046447 的基因进行了sgRNA 设计。这2 个基因在全基因组的比对结果如图3 所示。
2.1.2 sgRNA 设计结果 对ENSSSCG0000004 3 0 5 8 和E N S S S C G 0 0 0 0 0 0 4 6 4 4 7 位点进行sgRNA 设计(表5)。在sgRNA 及其互补序列末端分别加上与BpiI 酶切HP180_px330_GFP 质粒互补的黏性末端,由华大基因合成相应的Oligo。
2.2 sgRNA 体外切割结果
以野生型PEF DNA 为模板进行PCR 扩增,产物进行凝胶电泳分析后切胶回收得到sgRNA 体外转录模板。根据G e n e A r t T M P r e c i s i o n g R N ASynthesis Kit 试剂盒的体系与方法进行sgRNA 的转录模板合成(图4A) 与体外sgRNA 转录(图4B),转录产物经纯化后电泳验证条带完整性(长度100 bp),电泳结果与预期一致。
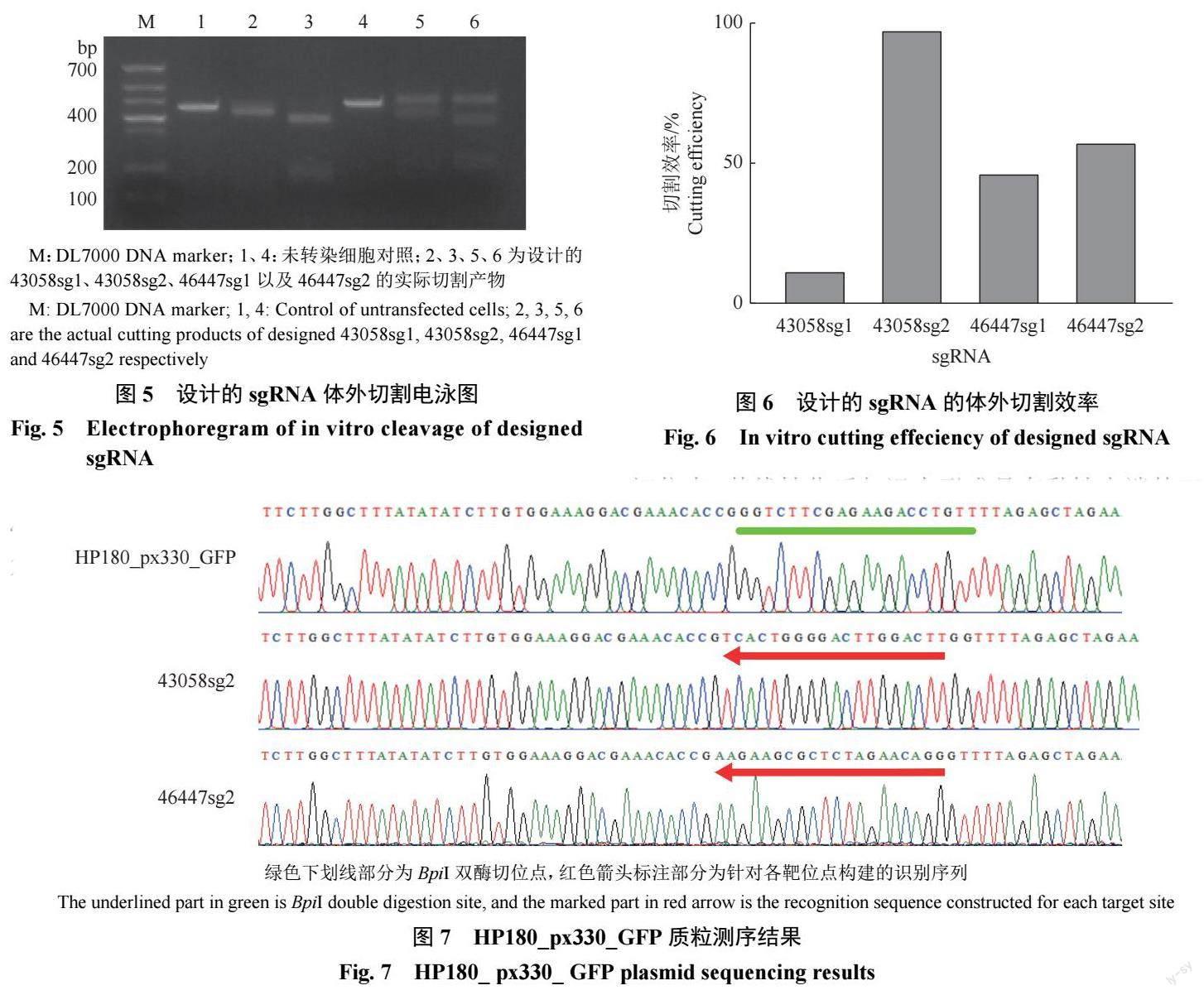
将扩增好的靶片段、sgRNA 按照一定比例与Cas9 混合,按照方法中的步骤进行反应得到切割产物,43058sg1 的理论切割产物长度为525 和91 bp,43058sg2 的理论切割产物长度为444 和172 bp,46447sg1 的理论切割产物长度为498 和159 bp,46447sg2 的理论切割产物长度为453 和204 bp。凝胶电泳分析表明43058sg2 与46447sg2 均达到了预期的切割效果,并且43058sg2 实现了接近完全的靶基因切割(图5)。灰度分析表明,46447sg2 的体外切割效率也达到了5 8 % (图6 ),最终我们选择43058sg2 和46447sg2 进行载体构建。
2.3 HP180_px330_GFP 载体构建结果
HP180_px330_GFP 骨架上有预留的BpiI 双酶切位点,其线性化后与退火形成具有黏性末端的双链sgRNA,在连接酶作用下形成闭合环状结构。其测序结果如图7 所示,结果与设计的sgRNA 结果一致,表明载体构建成功。
2.4 细胞性别鉴定及细胞转染检测
2.4.1 性别鉴定 对试验所用PEF 进行SRY 鉴定,电泳结果显示目的细胞为雄性(图8)。
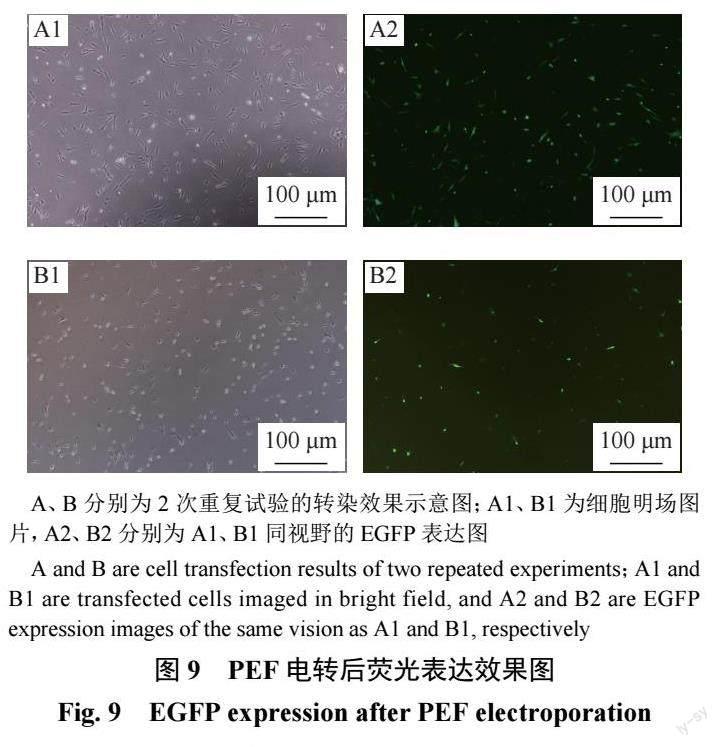
2.4.2 细胞转染 将构建的CRISPR/Cas9 载体通过细胞核转染导入雄性PEF 中。由于载体带有增强绿色荧光蛋白(Enhanced green fluorescentprotein,EGFP) 表达单元,可通过观察EGFP 的表达评估载体的转染效率。对已电转的细胞培养24 h后观察荧光信号,确定已构建好的载体成功电转进入细胞。结果(图9) 表明质粒已成功表达。
2.5 FACS 分选阳性细胞及单克隆鉴定结果
通过FACS 分选EGFP 阳性细胞,我们发现对照组与试验组GFP 信号存在显著差异,其中对照组未检测到GFP 信号,而阳性对照的细胞转染效率达到了71%,这与荧光观察结果类似。通过培养EGFP 阳性细胞,获得单细胞克隆团。接着我们对单克隆细胞团提取微量DNA,进行相应的SRY 检测,发现1/3 的细胞克隆未检测到SRY 信号(图10),显示这些细胞的Y 染色体存在缺失的可能。
2.6 实时定量荧光PCR 检测目的基因及SRY 基因
由于选用的目的基因在P E F 上并不表达mRNA,因此采用qPCR 检测目的基因(内参基因选择β-actin) 和SRY 基因的DNA 含量。我们抽提EGFP 阳性细胞的基因组DNA,进行qPCR 分析目的基因(43058 和46477) 和SRY 在DNA 水平的含量。qPCR 结果显示在进行靶基因敲除后,靶基因含量均显著降低。其中靶基因43058 和46447 分别下降了约20% 和30%(P<0.05),对应的SRY 也下降了7% 和11%。46447 敲除效果相对于43058 的基因敲除效果更为显著,不仅靶基因敲除效率较高,SRY 基因DNA 水平也下降较多(图11)。
2.7 核型分析结果
核型鉴定结果显示上述流式分选得到的EGFP 阳性细胞存在染色体缺失现象,表明设计的sgRNA 在細胞水平上能够实现整个Y 染色体的删除。在鉴定的12 个单细胞核型中,有1 个完全缺失Y 染色体(图12)。
3 讨论与结论
CRISPR/Cas9 近年来由于其基因编辑的灵活性和高效率而被广泛应用。其通过DSB 导致的非保守非同源末端连接修复途径造成目的基因的敲除效果,但是该种途径常常造成异常的插入缺失,甚至破坏染色体和基因组的完整性[24-26]。而以天然的(或定向的)DNA 模板的同源重组修复(Homologousrecombination repair,HDR) 途径则可以对目的基因位点进行精确修饰而产生意向的目的基因型[14, 27],本研究利用CRISPR 系统的高效切割能力对Y 染色体上多个重复基因位点进行切割,通过产生多重DSB 损伤基因组和DNA 修复的稳定性,引起Y 染色体降解。实现整条染色体删除是建立在CRISPR/Cas9 系统能对多个位点进行同时编辑的基础上的,虽然早期的研究已经证明能够产生大片段的缺失,但这对于整条染色体的消融还远远不够。2015年,Yang 等[ 2 8 ] 首先通过靶向内源性逆转率病毒(PERVs) 的62 个重复位点在猪上实现了PERVs 的全基因组降解;随后,研究人员进一步利用CRISPR/Cas9 对靶点切割的多重性,成功在ES 细胞、体内细胞和受精卵中删除了Y 染色体,并对其他常染色体(7 号、14 号) 及X 染色体进行了特异性消除[22-23]。
实现该技术的关键是在Y 染色体上寻找能够被sgRNA 特异识别的靶向序列,这些序列在其他常染色体或X 染色体上不能被识别,通过对其进行多重切割从而实现整条染色体的消融。因此在选择靶点时,单一数据库的比对结果不能很好地反映个体染色体组的真实情况,最终选择靶基因是在Ensembl比对的基础上重新与NCBI、Sanger 测序结果进行比对,选择多个数据库共有序列并与个体基因组序列结合,增加靶点的可靠性。
不同于一般的sgRNA 效率检测采用T7E1 酶切和测序验证,由于本试验靶点为Y 上的多拷贝重复序列,这些片段在Y 上广泛分布且保守性不强,可能会存在一些SNP 或者突变,导致扩增未编辑前的靶基因产物时出现套峰现象,和编辑后的测序结果无法区分。因此在这里只能通过体外切割进行sgRNA 的效率检测。最终以ID 为ENSSSCG00000043058、ENSSSCG00000046447 的基因为靶点设计sgRNA,通过对Y 上不同靶点,即散在重复和簇状聚集的靶基因的打靶,探索了不同打靶位点对编辑效率的影响,结果表明簇状聚集的打靶效果要明显优于散在重复序列,这与Zuo 等[22] 和Adikusuma等[23] 的研究结果一致,其原因可能是长臂偶然断裂造成的染色体截断或易位导致的。另外由于我们选择的基因为非蛋白编码基因,所以在定量分析时最终选择以DNA 为模板。在检测靶点DNA 含量变化的同时我们也对SRY 含量进行了定量,结果均表明其表达量随靶基因降低而降低,虽然这种变化并不明显,但从侧面说明了整条Y 染色体降解的可能性。核型分析的结果也表明部分单细胞克隆在染色体水平确实存在着Y 染色体的丢失,表明通过多位点断裂造成整条染色体的缺失是可行的。
本研究通过寻找能够特异性识别Y 染色体的多拷贝重复序列设计sgRNA,并通过体外切割、qPCR、细胞转染、核型分析等手段证明了该系统在体外体内都有较高的切割效率,可成功实现整条染色体的消除,为未来染色体功能及动物性别控制研究提供了新的方向。
參考文献:
ODOHERTY A, RUF S, MULLIGAN C, et al. An aneuploidmouse strain carrying human chromosome 21with Down syndrome phenotypes[J]. Science, 2005,309(5743): 2033-2037.
XIAO A, WANG Z, HU Y, et al. Chromosomal deletionsand inversions mediated by TALENs and CRISPR/Cas in zebrafish[J]. Nucleic Acids Research,2013, 41(14): e141. doi: 10.1093/nar/gkt464.
LI L B, CHANG K, WANG P, et al. Trisomy correctionin Down syndrome induced pluripotent stem cells[J]. CellStem Cell, 2012, 11(5): 615-619.
ABRAM C L, ROBERGE G L, HU Y, et al. Comparativeanalysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice[J].Journal of Immunological Methods, 2014, 408: 89-100.
TADA M, MATSUMURA H, KUROSE Y, et al. Targetchromosomes of inducible deletion by a Cre/invertedloxP system in mouse embryonic stem cells[J]. ChromosomeResearch, 2009, 17(4): 443-450.
CHIANG J C, JIANG J, NEWBURGER P E, et al. Trisomysilencing by XIST normalizes Down syndrome cellpathogenesis demonstrated for hematopoietic defects invitro[J]. Nature Communications, 2018, 9: 5180.
JIANG J, JING Y, COST G J, et al. Translating dosagecompensation to trisomy 21[J]. Nature, 2013, 500(7462):296-300.
SANTIAGO Y, CHAN E, LIU P Q, et al. Targeted geneknockout in mammalian cells by using engineered zincfingernucleases[J]. Proceedings of the NationalAcademy of Sciences of the United States of America,2008, 105(15): 5809-5814.
KIM H, KIM J. A guide to genome engineering with programmablenucleases[J]. Nature Reviews Genetics, 2014,15(5): 321-334.
SPITZ F, HERKENNE C, MORRIS M A, et al. Inversion-induced disruption of the Hoxd cluster leads to thepartition of regulatory landscapes[J]. Nature Genetics,2005, 37(8): 889-893.
LOONSTRA A, VOOIJS M, BEVERLOO H B, et al.Growth inhibition and DNA damage induced by Cre recombinasein mammalian cells[J]. Proceedings of the NationalAcademy of Sciences of the United States ofAmerica, 2001, 98(16): 9209-9214.
GARNEAU J E, DUPUIS M, VILLION M, et al. TheCRISPR/Cas bacterial immune system cleaves bacteriophageand plasmid DNA[J]. Nature, 2010, 468(7320):67-71.
DELTCHEVA E, CHYLINSKI K, SHARMA C M, et al.CRISPR RNA maturation by trans-encoded small RNAand host factor RNase III[J]. Nature, 2011, 471(7340):602-607.
YOSHIMI K, KUNIHIRO Y, KANEKO T, et al. ss-ODN-mediated knock-in with CRISPR-Cas for large genomicregions in zygotes[J]. Nature Communications,2016, 7: 10431.
ZHENG Q, CAI X, TAN M H, et al. Precise gene deletionand replacement using the CRISPR/Cas9 system inhuman cells[J]. BioTechniques, 2014, 57(3): 115-124.
GILBERT L A, HORLBECK M A, ADAMSON B, et al.Genome-scale CRISPR-mediated control of gene repressionand activation[J]. Cell, 2014, 159(3): 647-661.[16]
方锐, 畅飞, 孙照霖, 等. CRISPR/Cas9 介导的基因组定点编辑技术[J]. 生物化学与生物物理进展, 2013, 40(8):691-702.
BURMA S, CHEN B P C, CHEN D J. Role of non-homologousend joining (NHEJ) in maintaining genomic integrity[J]. DNA Repair, 2006, 5(9/10): 1042-1048.
HARTLERODE A J, SCULLY R. Mechanisms ofdouble-strand break repair in somatic mammaliancells[J]. The Biochemical Journal, 2009, 423(2): 157-168.
GASIUNAS G, BARRANGOU R, HORVATH P, et al.Cas9-crRNA ribonucleoprotein complex mediates specificDNA cleavage for adaptive immunity in bacteria[J].Proceedings of the National Academy of Sciences of theUnited States of America, 2012, 109(39): E2579-E2586.
JINEK M, CHYLINSKI K, FONFARA I, et al. A programmabledual-RNA-guided DNA endonuclease in adaptivebacterial immunity[J]. Science, 2012, 337(6096):816-821.
ZUO E, HUO X, YAO X, et al. CRISPR/Cas9-mediatedtargeted chromosome elimination[J]. Genome Biology,2017, 18(1): 224. doi: 10.1186/s13059-017-1354-4.
ADIKUSUMA F, WILLIAMS N, GRUTZNER F, et al.Targeted deletion of an entire chromosome using CRISPR/Cas9[J]. Molecular Therapy, 2017, 25(8): 1736-1738.
FU Y, FODEN J A, KHAYTER C, et al. High-frequencyoff-target mutagenesis induced by CRISPR-Cas nucleasesin human cells[J]. Nature Biotechnology, 2013,31(9): 822-826.
ADIKUSUMA F, PILTZ S, CORBETT M A, et al. Largedeletions induced by Cas9 cleavage[J]. Nature, 2018,560(7717): E8-E9.
CULLOT G, BOUTIN J, TOUTAIN J, et al. CRISPRCas9genome editing induces megabase-scale chromosomaltruncations[J]. Nature Communications, 2019, 10:1136.
LAPINAITE A, KNOTT G J, PALUMBO C M, et al.DNA capture by a CRISPR-Cas9-guided adenine baseeditor[J]. Science, 2020, 369(6503): 566-571.
YANG L, G?ELL M, NIU D, et al. Genome-wide inactivationof porcine endogenous retroviruses (PERVs)[J].Science, 2015, 350(6264): 1101-1104.
【責任编辑 庄 延】
猜你喜欢
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
安徽农业科学(2017年35期)2017-05-30
老年医学与保健(2017年6期)2017-02-06
中学生理科应试(2016年4期)2016-11-19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中央民族大学学报(自然科学版)(2016年3期)2016-06-27
哈尔滨医药(2015年2期)2015-12-01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
奇闻怪事(2014年5期)2014-05-13
