
Fe-MOF催化芳烃C(sp2)-H胺化反应研究
2023-08-10 03:29洪文杰王乐
山东化工 2023年11期
洪文杰,王乐
(东华大学 材料科学与工程学院 纤维材料改性国家重点实验室,上海 201600)
含氮化合物已成为药物化学、制药、天然产物合成、有机材料和催化剂领域的关键组成部分[1]。其中,芳基胺是许多天然产物和合成化合物中的关键成分,具有重要的化学、生物和药用特性[2]。分子内脱氢释放H2实现C-H胺化是一种非常有效的结合C-H键和N-H键的方法。理想情况下,芳香族的C-N键可以直接从相应的芳烃和胺中构建,只产生H2作为副产物[3]。但是这类反应存在几个问题使得转变并不是直接的[4]。首先,动力学和热力学屏障的存在使得反应无法自发进行,芳烃中C-H键的低极性以及高的pKa值进一步促成了它们的低反应性,且在底物中存在的强度和酸度相似的C-H键,就会发生区域选择性的问题。其次,两个耦合化合物都具有亲核性,导致极性不匹配,进一步阻碍了反应的进行。因此长期以来,人们一直在努力开发高效的反应,将含氮基团引入芳烃分子。
自从过渡金属催化的交叉偶联反应发展后,现代有机合成取得了相当大的进展[5]。传统上将氨基引入芳烃中最重要的方法是亲电芳香族硝化,然后还原硝基[6]。然而,芳烃的硝化通常需要强酸性或氧化反应条件,这限制了其在具有敏感官能团的底物上的使用。过渡金属催化的C-N交叉偶联反应是一种通过C-N键形成将氮引入有机分子的通用方法,它扩大了合成范围,缩短了合成程序。利用过渡金属催化C-N键的形成的方法之一是Buchwald-Hartwig胺化反应类交叉耦合反应[7-8]。该反应在钯和碱的存在下,胺化合物与芳基卤素化合物发生交叉偶联反应,形成C-N键,得到胺的N-芳基化合物。但这种交叉耦合反应需要在胺化反应前引入卤化物基团,且在反应后生成卤化物盐等副产物[9]。采用C-H键的胺化是催化C-N键形成的另一种方法,无需预安装反应基团,直接使碳氢化合物的底物官能化[10]。但由于C-H键的键能较高,pKa较低,因此C-H键的活化仍有一定的难度。将无处不在的C-H键催化转变为具有高经济价值的C-N键,是合成含N分子有效的合成方案。胺类化合物能通过C-H键的活化从芳烃合成,是合成芳基胺化合物的最具步骤和原子经济性的方法之一。
分子间直接胺化反应是一种强大而有力的合成工具,因为不需要合成制备所需要的底物。形成C-N键的交叉脱氢偶联仍然是最理想的,因为它避免了预功能化步骤。考虑到催化剂的经济和环境效益,在C-H功能化过程中使用廉价过渡金属催化剂是一种有吸引力的策略。金属催化的C-H键直接胺化反应中已经使用了多种胺化试剂[11]。尽管非官能团前化的胺或酰胺是最理想的氮源,但需要外部氧化剂才能与这些反应物进行催化C-H胺化反应。在金属催化剂的作用下,从胺化试剂衍生而出的带有极化N-X键的化合物能够发生氧化裂解[12]。因此这类胺化试剂可以在不额外添加氧化剂的情况下,也作为内部的氧化剂使用。虽然在实现芳族C-H胺化方面有了重大突破,但在适用的芳族化合物范围内仍然存在严重的局限性。由于获得反应性普遍困难,许多报道的反应仅适用于具有催化剂配位基团或电子活化的芳香族化合物。因此,开发用于简单芳烃的C-H胺化反应和催化剂一直是该领域的一项艰巨任务。
在过去的研究中,晶态多孔MOF材料因其结构可设计性、功能可设计性以及较好的物理以及化学稳定性等,已经被广泛地研究并应用于非均相催化领域[13]。在催化方面的研究MOF的晶体结构决定了其主要侧重于非均相催化。由于MOF的组成和结构特征的结合,MOF被认为是非均相催化中最有前途和最通用的材料之一。MOF的催化活性可以归因于其各种物理化学性质,例如高比表面积、大孔径、足够的框架稳定性、尺寸和形状的可调节性、易于回收、浸出少等[14]。MOF的这些特性使得在高活性下又具有高选择性,具有均相以及非均相催化剂的整体优势,使其在非均相催化中更有吸引力。与均相催化剂相比,MOF非均相催化剂可以在催化反应结束后回收使用,这解决了均相催化剂难以回收使用等难题。与传统的微孔以及介孔材料相比,高度的孔隙率和广阔的比表面积以及有机和无机成分的变化性为MOF的结构和性质提供了很大程度的多样性。得益于MOF拥有的精确晶态结构,因此它在非均相催化方面对催化活性位点的研究变得明确可行。MOF在C-H键的催化活化方面具有几个特点,如催化活性位点分布均匀、能牢固固定活性位点、金属和配体具有可修饰性以及可用单晶衍射仪精确确定空间结构表征催化活性位置[15]。这些都有利于MOF催化C-H键活化机理的研究。在数不清的MOF材料中,铁基金属有机框架具有结构多样性、毒性低、稳定性好、功能设计等诸多特性,在实际应用中具有极佳的潜力,受到了广泛的研究[16-17]。因此我们希望探索使用廉价铁基金属有机框架材料作为非均相催化剂,催化C-H键的活化反应。在此我们报道了使用廉价金属铁基金属有机框架材料Fe-MOF作为非均相催化剂,使用亲电胺化试剂作为胺源,以羟胺衍生物催化芳烃C-N键的形成反应。胺化试剂的N-O键相对较弱,在催化胺化过程中裂解时作为内部氧化剂的作用,从而避免了向反应混合物中添加外部氧化剂的需要。反应条件温和,无需对底物预安装导向官能团,成功实现简单烷烃取代的芳烃化合物C(sp2)-H胺化。
1 实验部分
1.1 Fe-MOF催化剂合成及表征
Fe-MOF的合成参考了先前文献报道的方法[18]。首先将四水合氯化铁(0.04 g,0.3 mmol)、1,3,5-苯三甲酸(0.1 g,0.45 mmol)溶解在N,N-二甲基甲酰胺(DMF,4.5 mL)和冰乙酸(1.5 mL)的混合溶液中。分散溶解均匀后,将混合反应溶液转移至水热釜中。采用溶剂热方法,于150 ℃烘箱中反应36 h。反应结束后,可以得到黄色块状晶体。将获得的晶体用DMF清洗3 d,每天清洗三次以除去未反应的原料。 用甲醇清洗3 d,每天更换三次新鲜甲醇溶液。将活化后的Fe-MOF在真空下干燥,获得深黄色块状晶体。粉末X射线衍射法是表征晶体结构强有力的手段之一,常用于晶体结构的物相分析。将使用水热法合成的Fe-MOF进行XRD分析测试,结果如图1所示。合成的Fe-MOF的衍射峰与文献中报道的图谱吻合。表明成功合成了晶态多孔材料Fe-MOF。

图1 Fe-MOF的XRD谱图
1.2 胺化试剂合成及表征
胺化试剂NsONH2MeOTf合成步骤如图2所示。
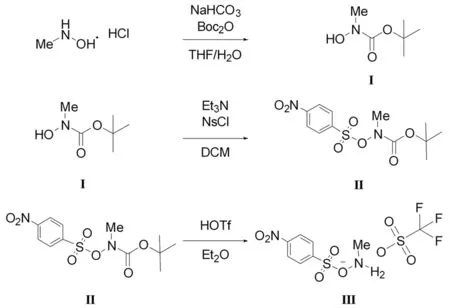
图2 胺化试剂合成反应式
将N-甲基羟胺盐酸盐(5.0 g,59.9 mmol,1.0 mol/L)溶解于四氢呋喃(100 mL)和水(10 mL)的混合溶液中,加入碳酸氢钠(10.0 g,119.0 mmol,2.0 mol/L)和二碳酸二叔丁酯(15.7 g,72.0 mmol,1.2 mol/L),室温下搅拌16 h。反应结束后,将反应液用水(50 mL)稀释,并用二氯甲烷(3×50 mL)萃取。合并的有机相用无水硫酸镁干燥,真空浓缩得橙色油状混合物。对获得的橙色油状混合物进行柱色谱分离纯化,在V(正己烷)∶V(乙酸乙酯)=3∶7洗脱剂下获得无色油状化合物Ⅰ(NMeOHBoc,6.7 g,45.6 mmol,76%)。1H NMR(600 MHz,Chloroform-d)δ3.17(s,3H),1.50(s,9H)。13C NMR(151 MHz,Chloroform-d)δ157.61,81.86,37.78,28.29。
将化合物Ⅰ(6.7 g,45.6 mmol,1.0 mol/L)溶解在二氯甲烷中(90 mL)。将溶液冷却至0 ℃后,加入三乙胺(4.6 g,45.6 mmol,1.0 mol/L),随后加入对硝基苯磺酰氯(10.17 g,46.0 mmol,1.0 mol/L)。将反应液升至室温并搅拌16 h。反应结束后,有机相用去离子水(3×50 mL)、碳酸氢钠溶液(3×50 mL)和饱和食盐水(3×50 mL)洗。合并的有机相用无水硫酸镁干燥,减压干燥获得灰白色固体Ⅱ(NsONMeBoc,12.6 g,38.0 mmol,83%)。无需进一步纯化即可进行下一步使用。1H NMR(600 MHz,Chloroform-d)δ8.44~8.39(m,2H),8.24~8.19(m,2H),3.31(s,3H),1.24(s,9H)。13C NMR(151 MHz,Chloroform-d)δ155.59,151.14,139.80,131.12,123.90,84.19,40.75,27.65。
将灰白色固体Ⅱ(12.6 g,38.0 mmol,1.0 mol/L)悬浮在乙醚(120 mL)中,将所得的悬浮液冷却至0 ℃,在剧烈搅拌下滴加三氟甲烷磺酸(5.7 g,38.0 mmol,1.0 mol/L)。添加完成后,使反应液升至室温并反应2 h。反应结束后,过滤固体,用冰乙醚(3×20 mL)洗涤,干燥得白色固体胺化试剂Ⅲ(NsONH2MeOTf,9.0 g,23.6 mmol,62%)。1H NMR(600 MHz,Acetonitrile-d3)δ9.54(s,2H),8.47~8.43(m,2H),8.25~8.20(m,2H),2.90(s,3H)。13C NMR(151 MHz,Acetonitrile-d3)δ152.71,140.48,131.60,125.66,39.69。19F NMR(565 MHz,Acetonitrile-d3)δ-79.50。
1.3 反应条件筛选
选取了均三甲苯作为反应条件优化实验的底物模型,以合成的试剂NsONH2MeOTf为胺化试剂,合成的Fe-MOF作为非均相催化剂,对反应溶剂、反应温度、催化剂用量等方面进行了条件筛选,如表1所示。首先筛选了在室温下反应溶剂对反应的影响,发现该N-甲基苯胺化反应高度依赖于1,1,1,3,3,3-六氟-2-丙醇(HFIP),在其余溶剂中皆无产物出现。推测是所使用的胺化试剂不溶于HFIP外的溶剂,观察反应现象,只有HFIP溶剂能充分溶解胺化试剂NsONH2MeOTf,其余溶剂不能溶解或者不完全溶解胺化试剂NsONH2MeOTf。初步确认反应溶剂后,调整反应温度,在40 ℃达到了94%的产率。进一步在40 ℃反应条件下优化催化剂Fe-MOF的使用量,当使用量为5 mg时,产率在93%,使用量15 mg时,产率在89%。继续提高催化剂的用量并不能较好提高反应产率。因此将催化剂Fe-MOF的使用量定为10 mg。

表1 Fe-MOF催化C(sp2)-H胺化反应条件优化
综上,Fe-MOF催化芳烃的C(sp2)-H胺化最优反应条件是:Fe-MOF(10 mg),溶剂为HFIP溶剂,40 ℃下反应3 h。
2 结果与讨论
在得到最优反应条件后,进一步对底物适用范围进行了拓展,底物范围拓展如图3所示。三烷基取代的均三甲苯(1)以及1,3,5-三乙基苯(2)获得了较高的产率,产率分别为94%,90%。对于简单的单烷基取代苯,反应具有中等的收率,对于甲苯(3)和乙苯(4)的产物,都只有单一的对位取代的N-甲基苯胺衍生物,产率分别为75%,56%。而单取代基是大位阻基团的叔丁基苯(5)则出现邻位和对位取代的两种N-甲基苯胺衍生物,产率为57%,邻位和间位产物比为11∶1。对于二烷基取代的间二甲苯(6),反应则表现出较高的产率和较好的选择性,产率为85%,且只有对应的单取代的产物。实验表明该体系具有高反应性以及优异的位点选择性。

图3 C(sp2)-H胺化底物范围扩展
N,2,4,6-tetramethylaniline(1):由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ6.89(s,2H),2.90(s,1H),2.80(s,3H),2.33(s,6H),2.29(s,3H)。13C NMR(151 MHz,Chloroform-d)δ144.95,131.37,129.59,129.52,35.66,20.62,18.22。
2,4,6-triethyl-N-methylaniline(2):由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ6.95(s,2H),2.93(s,1H),2.81(s,3H), 2.72(q,J=7.6 Hz,4H),2.64(q,J=7.6 Hz,2H),1.34~1.28(m,9H)。13C NMR(151 MHz,Chloroform-d)δ144.08,138.47,136.40,126.28,37.31,28.45,24.47,15.78,15.16。
N,4-dimethylaniline(3):由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.05~7.00(m,2H),6.57(d,J=8.4 Hz,2H),2.83(s,3H),2.27(s,3H)。13C NMR(151 MHz,Chloroform-d)δ147.25,129.80,126.59,112.73,31.21,20.49。
4-ethyl-N-methylaniline(4):由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.08(d,J=8.3 Hz,2H),6.62(d,J=8.3 Hz,2H),2.86(s,3H),2.60(q,J=7.6 Hz,2H), 1.24(t,J=7.6 Hz,3H)。13C NMR(151 MHz,Chloroform-d)δ147.39,133.20,128.57,112.65,31.11,28.01,16.10。
4-(tert-butyl)-N-methylaniline(5a):由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.28(dd,J=8.8,2.3 Hz,2H), 6.65~6.61(m,2H),2.87(s,3H),1.33(s,9H)。13C NMR(151 MHz,Chloroform-d)δ147.03,140.08,125.99,112.24,33.87,31.59,31.38。
3-(tert-butyl)-N-methylaniline(5b): 由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.19(t,J=7.9 Hz,1H),6.82~6.80(m,1H),6.69(t,J=2.1 Hz,1H),6.50(dd,J=8.0,2.4 Hz,1H),2.89(s,3H),1.35(s,9H)。13C NMR(151 MHz,Chloroform-d)δ152.26,149.13,128.91,114.72,110.14,109.34,34.66,31.01,30.91。
N,2,4-trimethylaniline(6):由快速色谱纯化系统分离得到黄色液体(洗脱剂:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.02(d,J=8.8 Hz,1H),6.95(s,1H),6.59(d,J=8.1 Hz,1H),3.46(s,1H),2.92(s,3H),2.30(s,3H),2.17(s,3H)。13C NMR(151 MHz,Chloroform-d)δ145.10,130.92,127.47,126.10,122.19,109.45,31.17,20.45,17.46。
3 结论
采用廉价金属基铁基MOF材料作为非均相催化剂,催化了简单芳烃的C(sp2)-H胺化反应,成功对简单芳烃实现直接胺化生成N-甲基苯胺类衍生物。使用高亲电的羟胺衍生物NsONH2MeOTf作为胺化试剂,既提供氮源,又包含容易离去的氧化基团,避免胺化反应中额外添加其他氧化剂。不需要在芳烃底物上预安装任何导向基团,即可在温和条件下获得一系列胺化产物,且反应具有优异的选择性。催化领域一项合乎演变的逻辑是将可溶性的均相反应转变为不可溶性的非均相反应。将MOF作为非均相催化剂催化有机反应的进行,并用于构建天然产物中间体有着巨大的应用价值。这为碳氢键的活化开辟了新的道路,并且对理解催化反应中的机理研究有着重要作用。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15
昆明医科大学学报(2021年8期)2021-08-13
应用化工(2021年4期)2021-05-20
科学家(2021年24期)2021-04-25
化工管理(2020年26期)2020-10-09
山东化工(2019年2期)2019-02-21
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
天然产物研究与开发(2016年11期)2016-06-15
橡胶工业(2015年9期)2015-08-29
