
二甲基姜黄素的合成工艺优化
2023-09-14 10:06张远卫李念光王晓波
药学与临床研究 2023年4期
张远卫,李念光*,吴 刚,王晓波,刘 飞*
1南京中医药大学药物化学教研室,南京 210000;2南京迈诺威医药科技有限公司,南京 210000
二甲基姜黄素(dimethyl curcumin,ASC-J9)是姜黄素的一种衍生物,化学名称为(1E,4Z,6E)-1,7-双(3,4-二甲氧基苯基)-5-羟基庚-1,4,6-三烯-3-酮,结构见图1。研究表明,二甲基姜黄素及其类似物对许多癌细胞表现出抗癌活性[1],例如通过破坏雄激素受体(androgen receptor,AR)与选择性AR 调节剂(AR 相关蛋白55 和AR 相关蛋白70)之间的相互作用,选择性地促进AR 降解[2]。此外二甲基姜黄素对结直肠癌[3]、雌激素依赖性乳腺癌细胞[4]、肝癌细胞[5]都有良好的抑制增殖作用。LIN TH 等[6]评估了目前使用的抗雄性激素比卡鲁胺(Casodex)、恩扎鲁胺(MDV3100)以及新开发的抗AR 化合物ASC-J9®和隐丹参酮对前列腺癌细胞生长和侵袭的影响,临床实验结果显示用ASC-J9®靶向雄激素受体比用比卡鲁胺或恩扎鲁安靶向雄激素的抗前列腺癌转移效果更好。

图1 二甲基姜黄素的合成路线Ⅰ
目前常用的二甲基姜黄素的合成工艺路线有两种。路线Ⅰ(图1):QIU X 等[7]用乙酰丙酮和氧化硼(B2O3)反应,制备硼络合物。将所得硼络合物与藜芦醛、除水剂硼酸三丁酯加入乙酸乙酯中,70℃下搅拌0.5、1 h 内滴加催化剂正丁胺,70℃下搅拌24 h。最后用浓度为1 mol·L-1的HCl 水溶液将体系pH 调至5,60℃搅拌0.5 h,静置分层,收集有机相,浓缩后再用乙酸乙酯(EtOAc)重结晶得到产物。尽管收率能达到90%,但该条路线反应溶剂需求量较大,反应时间较长;后处理需要通过萃取、浓缩等步骤,操作繁琐,会大大增加生产成本,不利于工业化生产。
路线Ⅱ(图2):SOMSAKEESIT LO 等[8]以姜黄素作为原料,四氢呋喃(THF)为溶剂,加入碳酸钾搅拌10 min 后加入碘化钾,回流搅拌3~5 h 制备二甲基姜黄素。路线Ⅱ较路线Ⅰ操作简单,但二甲基姜黄的收率仅有59%,另有副产物a:(1E,4Z,6E)-1-(3,4-二甲氧基苯基)-5-羟基-7-(3-羟基-4-甲氧基苯基)庚-1,4,6-三烯-3-酮、b:(E)-1,7-双(3,4-二甲氧基苯基)-1,5-二羟基-4-甲基庚-6-烯-3-酮产生,后处理需要进行柱层析分离产物,适用于实验室小试,不适于工业生产。

图2 二甲基姜黄素的合成路线Ⅱ
除了上述方法之外,还有其它方法可用于二甲基姜黄素的合成。LAALI KK 等[9]使用改进的微波辅助方法(图3),将curcuminoid-BF2 复合物和草酸钠加入到配备磁子的清洁干燥的微波小瓶中,再加入甲醇水溶液,密封后在140℃下照射6 min。冷却后,转移至圆底烧瓶中除去甲醇,加入去离子水形成沉淀。过滤,洗涤固体并干燥,可得到纯度高达98%的姜黄素类产物,且二甲基姜黄素的收率高达94%。同样是微波照射条件,ELAVARASAN S 等[10]向硼硅烧杯中加入藜芦醛、乙酰丙酮和碘浸渍的纳米中性氧化铝,搅拌混合均匀,然后在微波炉中以160 W照射40~110 s,冷却后,用乙酸乙酯(EtOAc)萃取,并过滤除去催化剂,乙酸乙酯提取物用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏得产物,产率同样达到80%。两种方法的产物收率高,反应条件为微波照射,对环境友好。但是无论是微波照射或者140℃的高温条件在工业化生产上均不适用。
综合上述所有反应路线,本研究选择路线I 进行工艺优化:如图4,第一步制备硼络合物的反应中,选择醋酸异丙酯作为反应溶剂,一定程度上提高乙酰丙酮的转化率。在后一步制备二甲基姜黄素的反应中,更换反应溶剂为N,N-二甲基乙酰胺(DMA)。通过溶剂的筛选可达到三个目的:①增大对底物的溶解度,减少溶剂体积,节约生产成本;②选择高沸点溶剂,提高反应温度至80℃,缩短反应时间至3 h;③提高每步的反应收率,且在后处理中产物可直接在反应淬灭后析出,免于萃取、分层、浓缩等步骤,操作更简单,更适合工业生产。
1 材 料
1.1 仪器
RW20 机械搅拌器(德国IKA 公司);电子天平(上海舜宇恒平科学仪器有限公司);AV400 核磁共振仪(400 Hz,德国Bruker 公司);旋转蒸发仪(德国Heidolph 公司);Arc 高效液相色谱仪配PDA2998紫外检测器和SQ2 单四级杆检测器,Empower 3 软件(美国Waters 公司)。
1.2 试剂与试药
氧化硼、正丁胺(上海麦克林化学试剂有限公司);乙酰丙酮、醋酸异丙酯、N,N-二甲基甲酰胺(DMF)、DMA、N-甲基吡咯烷酮(N-methylpyrrolidone,NMP)、藜芦醛、硼酸三丁酯(上海安耐吉试剂有限公司);石油醚、盐酸、无水硫酸钠、乙酸乙酯(上海沪试实验室器材股份有限公司)。
2 方 法
2.1 乙酰丙酮硼络合物的合成
称取乙酰丙酮(100.1 g,1.0 mol)和醋酸异丙酯(1000 mL)加入至3 L 的三颈瓶中,搅拌的条件下加入氧化硼(48.7 g,0.7 mol),维持反应体系温度为70℃,搅拌3 h,有大量白色固体析出。静置降温至25℃,抽滤,并用石油醚(100 mL)洗涤滤饼两次。30℃真空干燥1.5 h,得白色粉末状固体(127.4 g),无须纯化直接用于下一步反应。
2.2 二甲氧基姜黄素的合成
称取藜芦醛(332.3g,2.0mol),硼酸三丁酯(460.3g,2.0 mol)于2 L 三颈瓶中,加入DMA(500 mL),维持体系温度80℃,搅拌0.5 h。待体系溶清后,一次性加入“2.1”中制备的乙酰丙酮硼络合物(127.4 g),搅拌10 min。然后缓慢滴加正丁胺(14.6 g,0.2 mol),滴毕在此温度下搅拌反应3 h。薄层色谱监测反应转化完全,加入预热至60℃的HCl(0.3 mol)水溶液108 mL,降温至60℃搅拌0.5 h,再补加水(1000 mL)进行结晶。当内温55℃时,加入晶种(2.0 g)。降至室温后,在冰浴的条件下继续搅拌3 h,析出大量固体。抽滤,得粗品黄色固体377.0 g,含量98.6%,收率95.1%。
粗产物用800 mL 的DMA:H2O(1∶1.5,V/V)体系重结晶,过滤得淡黄色固体365.9 g,熔点129~131℃,总收率92.3%,纯度99.2%。MS(ESI+)m/z:397.1 [M+H]+;1H-NMR(400 MHz,DMSO-d6)δ 7.59(d,J=15.8 Hz,2H),7.36(d,J=2.0 Hz,2H),7.27(dd,J=8.4,2.0 Hz,2H),7.02(d,J=8.4 Hz,2H),6.84(d,J=15.9 Hz,2H),6.12(s,1H),3.83(d,J=8.6 Hz,12H)。
3 合成工艺优化
在证明该条路线可行的情况下,为了提高反应的收率,以10 g 乙酰丙酮作为原料,醋酸异丙酯为溶剂制得乙酰丙酮硼络合物,然后从反应溶剂、反应温度、淬灭所需的盐酸和水的用量及结晶所需水的用量等几个方面优化第二步反应。
3.1 反应溶剂的筛选
操作方法参考上述工艺,在其他条件不变的情况下,对反应溶剂进行考察,结果如表1 所示。
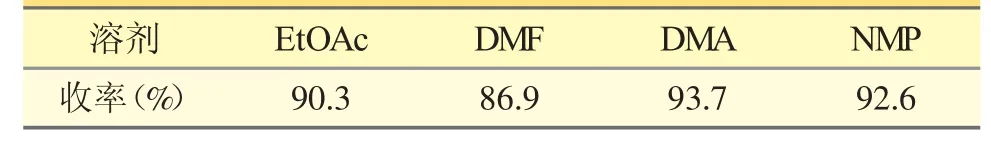
表1 不同溶剂对产物收率的影响
从表1 可得,以乙酸乙酯、DMA、NMP 反应溶剂均有较好的收率,收率≥90%。但乙酸乙酯作为反应溶剂后续处理需要进行萃取、浓缩步骤,且淬灭过程中,属于非均相体系,需要加入大量盐酸才能保证反应彻底淬灭,不适合工业生产。而DMA 或者NMP 作为反应溶剂,与水互溶,在淬灭过程中不仅能减少盐酸的用量,且产物能直接从体系中析出,操作更加简便。综合考虑,选择DMA 作为反应溶剂更优。
3.2 淬灭所需的酸用量筛选
在其他条件不变的情况下,对淬灭反应所需的酸的量进行考察,结果如表2 所示。
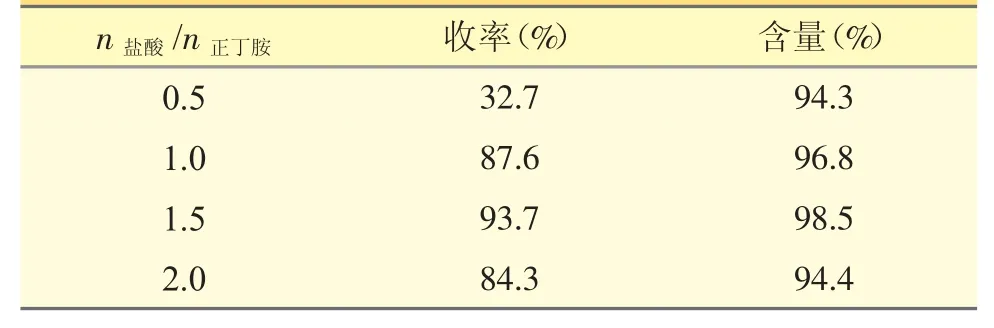
表2 不同盐酸的量对产物收率及含量的影响
由表2 可知,在进行后处理时,盐酸物质的量对反应收率影响较大。盐酸用量较少时,二甲基姜黄素未能从络合体系中完全解离出;盐酸用量高时,高浓度的酸对已解离出的二甲基姜黄素产生影响,使部分分解,均导致收率降低。当n盐酸/n正丁胺=1.5时,二甲基姜黄素基本解离析出,收率≥93%。
3.3 结晶所需水的用量筛选
在其他条件不变的情况下,对结晶所需的水的体积进行考察,结果如表3 所示。

表3 不同体积的水对产物收率及含量的影响
由表3 可得,在同等条件下,当V水/V溶剂≤2.0 时,后处理中补加的水的体积与收率成正比,含量成反比;当V/V >2.0 时,收率及纯度会同时下降。经考察当V/V=2.0 时,能得到较好的收率与纯度。
3.4 合成二甲基姜黄素的反应温度筛选
在其他条件不变的情况下,对反应温度进行了考察,结果如表4 所示。

表4 不同反应温度对产物得率及纯度的影响
由表4 可得,当反应温度分别为80、90、100℃时,转化率≥90%。但温度为100℃时,产物的纯度降低,可能是发生副反应,导致副产物增多,故80℃是较优的合成二甲基姜黄素的反应温度,此时反应收率为93.7%。
4 结果与讨论
本研究参考姜黄素的合成文献[11],对二甲基姜黄素的合成工艺进行优化,更换反应溶剂体系为DMA,反应温度80℃,反应时间3 h。同时优化了后处理条件,酸淬灭当量为碱当量的1.5 倍,补加2 倍溶剂体积的水,并添加晶种辅助结晶。
相较于路线Ⅰ,该法制备规模扩大到百克级别,收率92.3%,纯度>99%。反应条件温和,产物能直接从反应体系中析出,免除路线Ⅰ中萃取、浓缩等繁琐步骤。同时,溶剂体积大大降低,缩短反应时间,节约了生产成本,适合放大生产。
猜你喜欢
分子催化(2022年1期)2022-11-02
上海计量测试(2020年1期)2020-03-18
浙江大学学报(工学版)(2016年2期)2016-06-05
食品界(2016年4期)2016-02-27
大连工业大学学报(2015年4期)2015-12-11
环境科技(2015年1期)2015-11-08
云南中医学院学报(2015年2期)2015-07-31
化学反应工程与工艺(2015年1期)2015-04-16
中国塑料(2014年4期)2014-10-17
应用化工(2014年4期)2014-08-16
