
儿童线粒体脑肌病临床特点分析
2023-09-20 09:01卓木清李小晶彭炳蔚朱海霞郑可鲁高媛媛吴文晓吴汶霖陈宗宗陈文雄曹彬彬
临床儿科杂志 2023年9期
卓木清 李小晶 彭炳蔚 朱海霞 田 杨 郑可鲁 高媛媛 吴文晓 吴汶霖 陈宗宗 陈文雄 曹彬彬
广州市妇女儿童医疗中心神经内科(广东广州 510623)
线粒体脑肌病(mitochondrial encephalomyopathy,ME)是一种因线粒体基因(mitochondrial DNA,mtDNA)或细胞核基因(nuclear DNA,nDNA)变异导致呼吸链氧化磷酸化异常,从而引起能量高需求的神经系统及肌肉受累的遗传代谢性疾病。线粒体疾病可在任何年龄起病,但儿童往往为急性起病[1]。由于ME具有临床异质性,临床表现多样化,缺乏特异性,易被误诊而延误治疗。因此,本研究旨在探讨儿童ME的临床特点,以进一步提高对儿童ME的认识并做出早期诊断。
1 对象与方法
1.1 研究对象
回顾性分析2015 年1 月至2022 年8 月在广州市妇女儿童医疗中心神经内科诊治的儿童ME 患者的临床资料。纳入标准:①年龄28 d~18岁;②符合ME诊断标准,即基因诊断明确或按照Morava线粒体疾病的评分系统(Neurology 2006)[2]≥8分。排除标准:其他病因所致的脑肌病或多器官多系统的疾病。
1.2 方法
1.2.1 临床资料收集 通过医院电子病历系统回顾性收集入组ME 患儿的临床资料、神经系统影像学、肌肉活检及基因检查结果。
1.2.2 实验室检查 采集患儿6~8 mL外周血进行血乳酸(静息时)、血气分析、血酮体乳酸丙酮酸比值、血氨、生化功能、血氨基酸及酰基肉碱分析,存在长节段脊髓病变的患儿需检测血中枢神经脱髓鞘3项抗体,即抗水通道蛋白4(AQP4)抗体IgG、抗髓鞘少突胶质细胞糖蛋白(MOG)抗体IgG、抗髓鞘碱性蛋白(MBP)抗体IgG。征得患儿监护人的知情同意,留取患儿脑脊液3~5 mL进行脑脊液常规、生化、寡克隆、常见病原学(单纯疱疹病毒、EB病毒、肠道病毒、流感病毒、支原体、衣原体)及脑脊液酮体乳酸丙酮酸比值检测。
1.2.3 基因检测 本研究征得患儿监护人的知情同意,并经广州市妇女儿童医疗中心伦理委员会批准(穗妇儿科伦批字[2019]第40101号),采集患儿外周血细胞2 mL 或外周组织黏膜及其父母外周血细胞2 mL,应用二代测序技术进行全外显子及线粒体基因组测序。
1.3 统计学分析
采用SPSS 25.0 统计软件进行数据分析。计量资料符合正态分布的以均数±标准差表示,非正态分布的以中位数(M)(P25~P75)或M(全距)表示,组间比较采用秩和检验。计数资料以例数(百分比)表示。以P<0.05为差异有统计学意义。
2 结果
2.1 一般临床资料
纳入ME患儿37例,1例Morava线粒体疾病系统评分为8分,但其临床表型及基因型考虑Hartnup病而被排除。最终纳入患儿36 例。男22 例、女14例,起病年龄6.8(2.1~10.8)岁。15例(41.7%)患儿起病年龄<6 岁,男∶女为9∶6,Leigh 综合征(Leigh syndrome,LS)多见(7/15,46.7%);而起病年龄>6 岁常见于ME 伴高乳酸血症和卒中样发作(mitochondrial encephalopathy,lactic acidosis and stroke like episodes,MELAS)(12/21,57.1%),男∶女为13∶8。
患儿既往史:生后出现运动发育迟缓、体重增长缓慢各5例,消化道出血行十二指肠切除术1例、生后低血糖1 例、反复腹痛1 例、生长激素缺乏1 例及肠梗阻1例。家族史:2例患儿有同胞夭折史,1例为2个同胞(1个哥哥+1个姐姐)均在新生儿期死亡;另外1例为3个同胞(1个哥哥+2个姐姐)均在1岁左右发热后死亡,死因不明。
2.2 临床症状和表型
首发症状依次为卒中样发作24例(66.7%)、运动耐力下降7例(19.4%)、上眼睑下垂3例(8.3%)及智力发育迟缓2 例(5.6%)。病程中出现的神经肌肉系统症状包括:癫痫发作17例、头痛11例、瘫痪11例、共济失调10例、上眼睑下垂7例、精神/运动发育迟滞7 例、视物模糊6 例、意识障碍3 例、精神行为异常2 例。其他非神经肌肉系统症状包括发热(7 例)、呕吐(6 例)、体重增长缓慢(5 例)、腹痛(2例)、尿潴留(1例)、肠梗阻(1例)及镇静后呼吸衰竭(1例)。临床表型包括:MELAS 16例,LS 11例,肌阵挛癫痫伴破碎红纤维(myoclonus epilepsy with ragged-red fibers,MERRF)3例,卡恩斯-塞尔综合征(Kearns-Sayre syndrome,KSS)2例,不可归类4例。见表1。
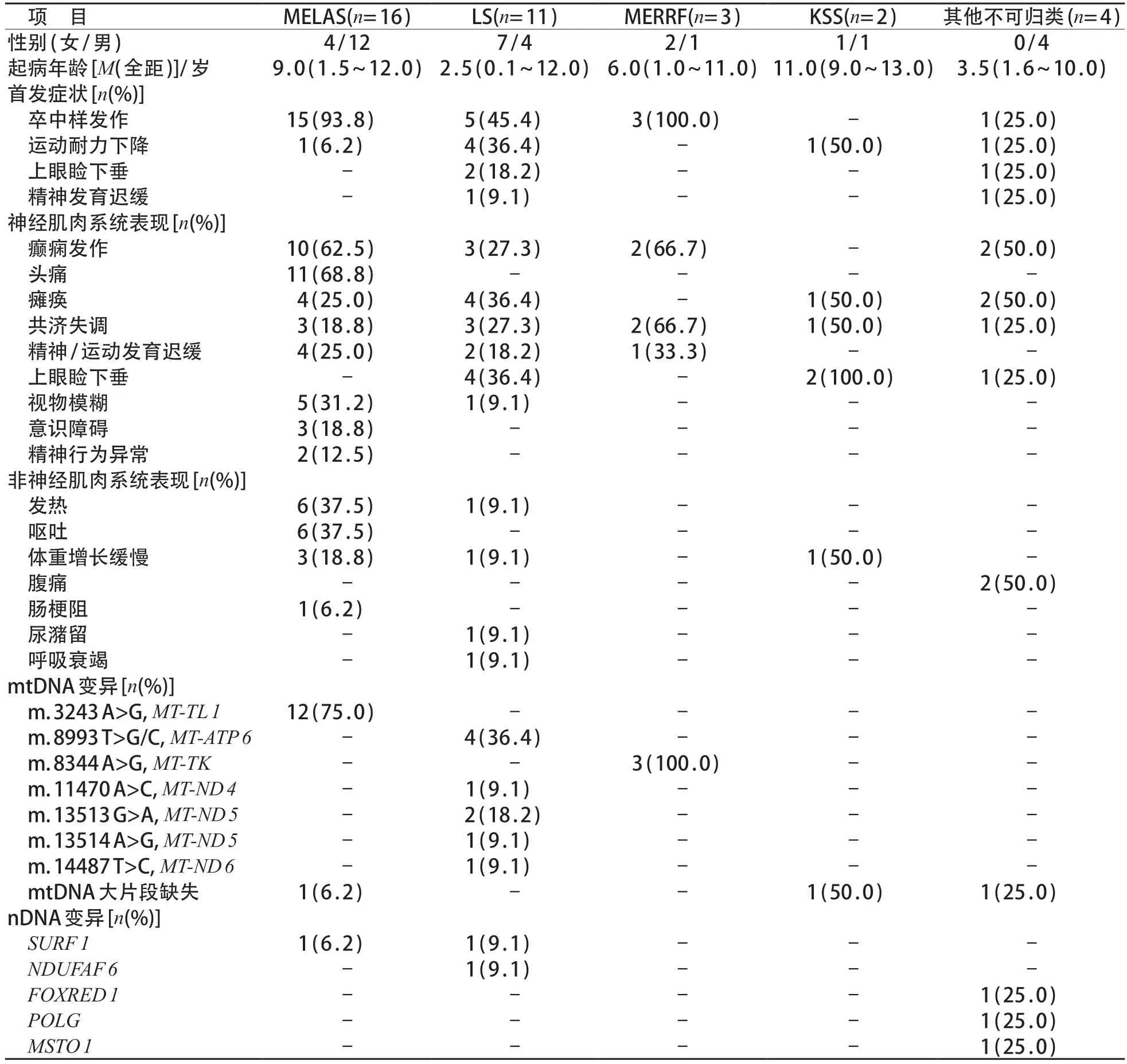
表1 ME患儿36例临床资料
3 例以上眼睑下垂为首发症状的患儿曾在外院行新斯的明试验,结果为阳性,被诊断为眼肌型重症肌无力,1例予类固醇激素口服治疗1年后好转停药,进展期以双下肢瘫痪为主要症状,本院查重症肌无力抗体显示阴性;1例予口服溴吡斯的明1年后好转停药,其后出现频繁癫痫发作至本院就诊;1例予溴吡斯的明联合类固醇激素治疗2年有好转但反复,本院查重症肌无力抗体显示阴性。
2.3 实验室检查
71.4 %(25/35)患儿静息时血乳酸有不同程度升高,95.2%(20/21)脑脊液乳酸升高,且静息时血乳酸与脑脊液乳酸之间差异有统计学意义(Z=3.29,P=0.001)。66.7%(14/21)脑脊液丙酮酸升高,100%(21/21)脑脊液乳酸丙酮酸比值均大于20,15.1%(5/33)血肌酸激酶升高,33.3%(11/33)血乳酸脱氢酶升高。见表2。

表2 ME患儿实验室检查
2.4 磁共振成像检查
35 例患儿完善头颅磁共振成像(MRI)检查,4例头颅MRI 检查未见颅内病变。31 例患儿的头颅MRI显示病变部位为:额叶7例(22.6%)、顶叶15例(48.4%)、颞叶7例(22.6%)、枕叶14例(45.2%)、基底节13例(41.9%)、丘脑8例(25.8%)、小脑7例(22.6%)、脑干10例(32.3%)、胼胝体3例(9.7%),其中伴有大脑或小脑半球轻度萎缩分别为2 例和6例。
9例行头颅磁共振波谱(MRS)检查均提示病变区N-乙酰天门冬氨酸(NAA)峰下降,NAA/肌酸(Cr)降低,乳酸(Lac)峰明显升高。4例患儿同时完善全脊髓MRI检查,其中2例结果未见异常;1例颈、胸、腰段脊髓全段见斑片状异常信号,1 例病变部位为颈、胸、腰段多发脊髓异常信号,均为T1 加权像呈低信号,T2加权像呈高信号。见图1。
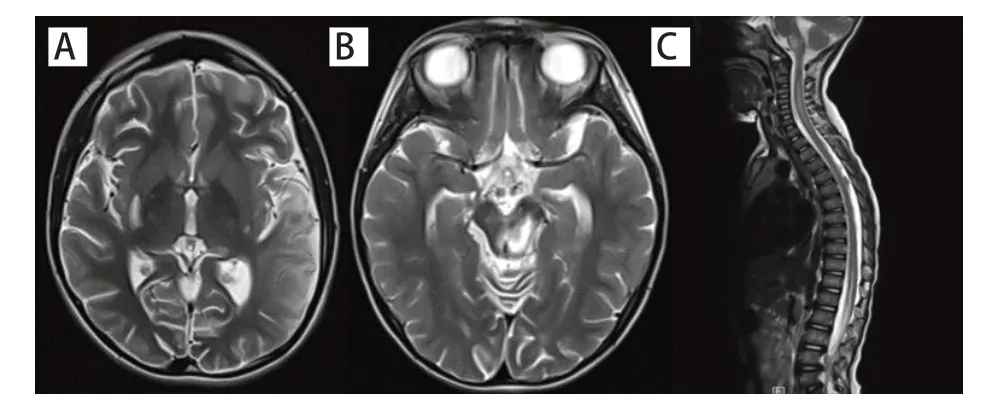
图1 ME 患儿头颅及脊髓MRI 结果
2.5 肌肉活检
4 例患儿行肌肉活检,苏木精-伊红(HE)染色可见肌纤维大小轻度不均,少许散在分布的小角状萎缩肌纤维,可见少许坏死、吞噬、再生肌纤维及散在嗜碱性肌纤维(图2 A)。2 例改良Gomori 三色法(MGT)染色可见散在破碎红纤维,未见镶边空泡(图2B);2例琥珀酸脱氢酶(SDH)染色见破碎蓝纤维及强反应血管(SSV)(图2 C),2 例细胞色素C氧化酶(COX)双染可见深染纤维及散在阴性肌纤维(图2D)。
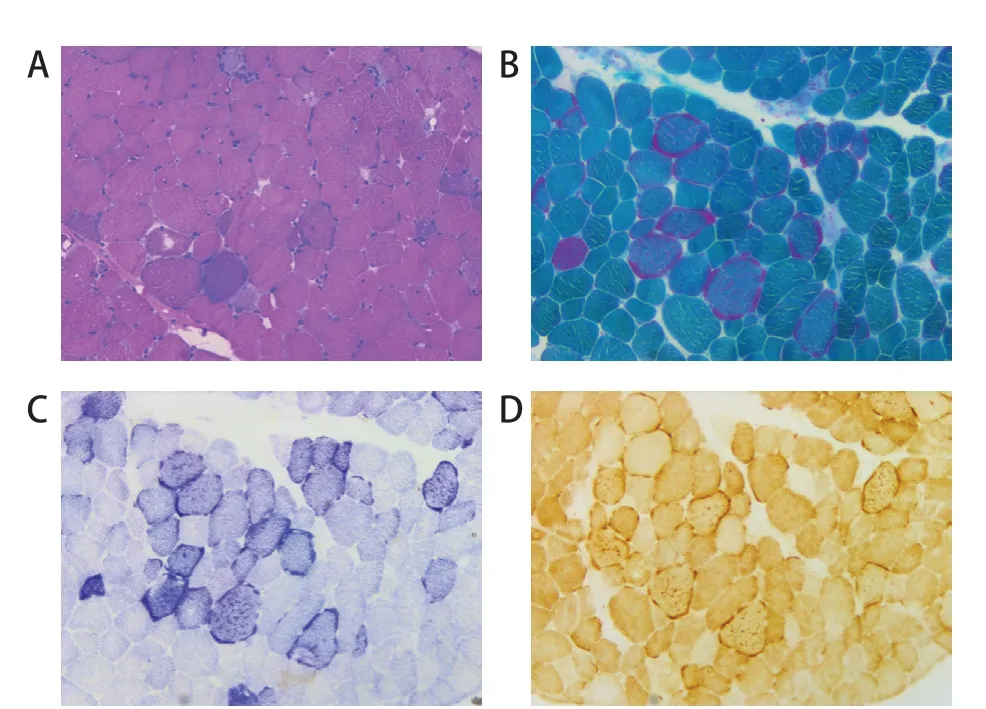
图2 ME 患儿光镜下横纹肌组织观察
2.6 基因测序结果
34 例患儿行全外显子及线粒体基因组测序,1 例阴性,33 例阳性患儿中mtDNA 变异27 例(81.8%),以MT-TL1∶m.3243A>G变异居多(12例,36.4%),其中1 例患儿外周血细胞基因测序结果为阴性,但其口腔黏膜成纤维细胞基因测序结果为mtDNA片段缺失;6例(18.2%)为nDNA变异,分别为SURF12例及NDUFAF6、FOXRED1、POLG、MSTO1各1例。见表1。
3 讨论
线粒体是ATP合成的主要场所,超过90.0%的细胞能量通过氧化磷酸化产生,目前已知mtDNA或nDNA 变异可导致呼吸链氧化磷酸化异常[3],从而导致能量高需求系统受累而出现多样化临床症状,这一类疾病被称为线粒体疾病,同时累及神经系统及肌肉的线粒体疾病称为ME。ME 可在任何年龄起病,本研究的起病年龄为6.8(2.1~10.8)岁,呈偏态分布,MELAS 起病年龄比LS 年长,男性多见(75.0%),这与相关临床分析的结果一致[4]。儿童ME临床表现为癫痫、卒中样发作、头痛、共济失调、精神运动发育倒退、智能倒退、精神行为异常以及同时出现累及多系统的症状,如耳聋、视力下降、肝功能异常等[1],其起病往往为急性,常见的代谢应激因素为炎症、饥饿、手术或镇静药物等。本研究中7例患儿急性起病时有发热感染病史,1例2月龄婴儿因需完善听觉脑干诱发电位检查而进行镇静,随后出现急性呼吸衰竭,急查血乳酸高达14.6 mmol/L,线粒体基因结果显示MT-ATP6m.8993T>G,因此对于镇静后出现急性呼吸衰竭、血乳酸升高且无相关呼吸道系统疾病的小婴儿,需高度警惕ME可能性。MELAS是ME最常见的临床表型,本研究中44.4%诊断为MELAS,这与相关研究报道的儿童线粒体疾病MELAS 比例约为46.7%[4]一致。卒中样发作是MELAS 的核心症状,本研究卒中样发作临床表现主要为癫痫发作、头痛及瘫痪。癫痫发作是ME常见的临床表现,本研究中约有半数患者出现癫痫发作(47.2%),与既往文献报道相符[5]。反复的卒中样发作还可导致智力进行性下降。本研究4 例患儿在小学3 年级以后学习成绩明显下降,且自诉记忆力明显下降,可能与反复卒中样发作导致皮层萎缩及多发软化灶改变有关。ME 具有临床及基因异质性,本研究POLG基因c.2558G>A(p.R853Q)、c.2890 C>T(p.R 964 C)复合杂合变异同卵双胎兄弟的临床表现存在差异,先证者主要临床表现为癫痫发作、肢体乏力,但其同胞哥哥至今暂无癫痫发作[6]。他们自幼均有反复腹痛、易疲劳、体型瘦小、生长发育落后于正常同龄儿的非特异性临床表现,且有3 个同胞均在1 岁发热后夭折,提示有同胞异常夭折史的患儿出现体型瘦小、运动耐力下降或生长发育落后,需注意排查儿童ME可能。此外,本研究3例以眼睑下垂为首发症状,曾在外院诊断为眼肌型重症肌无力,2例口服溴吡斯的明或类固醇激素后临床症状好转,疾病进展期时分别以双下肢瘫痪及频繁癫痫发作为主要症状,外周血乳酸呈不同程度升高,波动在2.2~7.1 mmol/L,基因结果为MT-ND5m.13513G>A和m.13514A>G,临床表型均为LS。另外1例治疗后虽有好转但反复,基因结果为mtDNA大片段缺失。众所周知,ME与重症肌无力具有不少相似的临床表现,比如上睑下垂、眼肌麻痹、易疲劳等。有研究提出针对非典型和常规治疗欠佳的重症肌无力需注意ME 可能,并结合国内外文献报道提出线粒体ND5基因很可能是伴眼睑下垂的LS 患儿的高发变异基因的猜想[7]。本研究约30.5%的患儿出现头痛,而头痛伴呕吐作为非特异性症状可见于众多疾病,但其也是卒中样发作的重要临床症状之一。由于ME临床表现多样化,缺乏特异性,易被误诊为脑炎、脑梗死、重症肌无力等,因此相关的辅助检查对进一步明确诊断非常重要。
中枢神经系统影像学检查对儿童ME 诊断及评估起到重要作用[8]。ME在MRI上有7种基本中枢神经系统异常,包括弥漫性小脑萎缩、脑萎缩、皮质下结构(基底节、脑干、小脑)对称性改变、大脑皮质和皮质下白质不对称信号改变、白质脑病以及视神经和脊髓的对称信号改变[9]。ME 的MRI 病灶常见于皮质及皮质下灰质,且病变范围多与脑动脉灌注供血区分布不一致,多位于顶枕颞叶、深部灰质核团,比如基底节、丘脑、脑干等[10]。本研究中31 例头颅MRI显示病变部位多位于顶叶(48.4%)、枕叶(45.2%)、基底节(41.9%)及脑干(32.3%),与既往文献报道[10]相似。研究表明,MELAS的头颅MRI病变区具有高灌注及低代谢特点[11],高灌注是由于乳酸堆积导致血管舒张所致,而低代谢是由于细胞毒性水肿后造成皮层萎缩及多发性软化灶所致。因此,作为可无创检测脑细胞代谢产物的MRS对疑似ME的患者具有诊断作用[9],尤其在急性期。本研究中头颅MRS均可见病变区NAA峰下降,NAA/Cr降低,Lac峰明显升高,同时监测患儿外周血及脑脊液乳酸可有不同程度升高,提示患儿组织能量代谢异常,糖酵解增多。本研究中约71.4%患儿静息时外周血乳酸有升高,而脑脊液乳酸升高可高达95.2%,两者间差异具有统计学意义(P<0.05)。并且,本研究中有8 例患儿外周血静息时乳酸为正常,而脑脊液乳酸却有不同程度升高(2.5~8.9 mmol/L),更进一步说明脑脊液乳酸比外周血乳酸具有更高敏感性。此外,本研究有2 例LS 同时可见长节段脊髓节段信号改变。有研究回顾性分析119 例原发性线粒体疾病儿童脊髓MR 特点,最终入组只有33 例,58.0%脊髓MR可见脊髓异常信号,白质和/或灰质同时受累的脊髓MR 改变与视神经脊髓炎谱系疾病(NMOSD)、多发性硬化(MS)、抗MOG抗体疾病相似[12]。因此,患儿脊髓 MRI出现长节段异常信号时,也需鉴别ME可能。
儿童ME 常见的骨骼肌受累表现为运动耐力下降、肢体乏力,部分患儿外周血肌酶可出现不同程度升高。肌酸激酶是肌肉损伤最敏感的指标,而丙酮酸转化为乳酸是乳酸脱氢酶催化糖酵解的最后一步,因此肌酸激酶和乳酸脱氢酶可作为辅助诊断儿童ME 的生化指标。本研究中伴有血肌酸激酶或乳酸脱氢酶升高的比例为15.1% 和33.3%。但肌酶升高也可见于炎性肌病、感染性肌病、横纹肌溶解、肌营养不良等疾病,对儿童ME的特异性低,因此肌活检是诊断ME 的重要手段,破碎红纤维(RRF)是最典型的病理表现。但肌活检作为一种有创的诊断方法,患儿家属对此接受度偏低,因此本研究仅有4例患儿完善肌活检,2例可见典型的RRF改变。相对而言,患儿家属更愿意接受基因检查,且基因检查是目前诊断ME的金标准。文献报道约15.0%~30.0%的儿童线粒体疾病由mtDNA变异所致[1]。相关研究对我国儿童ME 进行临床分析,发现儿童ME mtDNA变异可高达62.5%及65.0%,其热点致病变异位点均为m.3243 A>G[13]。而本研究中mtDNA 变异却高达81.8%,推测可能与本研究纳入的病例数不多有关。值得一提的是,mtDNA变异在不同组织存在巨大差异,根据ME 的临床分型选择不同组织标本进行基因检测,可进一步提高确诊率[14]。本研究有1 例患儿考虑KSS,其外周血细胞基因测序结果为阴性,但其口腔黏膜成纤维细胞基因测序结果为mtDNA片段缺失。此外,本研究中MSTO1基因移码变异c.13delG(p.Ala5ProfsTer68)已被前期研究报道,丰富了ME基因变异数据库[15]。
综上所述,本研究中儿童ME 多见于学龄期儿童,以MELAS 常见,首发症状多为卒中样发作,部分LS首发症状可仅表现眼睑下垂,随着病程进展可出现运动耐力下降、癫痫发作,提示对于眼睑下垂患儿伴有静息时外周血乳酸升高,需注意LS 可能性。而且,本研究发现2 例LS 出现长节段脊髓病变,这与典型的LS神经影像学改变截然不同,提示针对长节段脊髓病变需鉴别ME可能性。
猜你喜欢
军事文摘(2022年8期)2022-11-03
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中华养生保健(2020年8期)2021-01-14
中国运动医学杂志(2016年3期)2016-07-10
腹腔镜外科杂志(2016年12期)2016-06-01
恋爱婚姻家庭·养生版(2016年2期)2016-02-17
河北科技大学学报(2015年6期)2015-03-11
癌变·畸变·突变(2014年1期)2014-03-01
中国中医药现代远程教育(2014年21期)2014-03-01
