
螺癸二烯酮合成方法学的研究进展
2023-10-31 07:39刘天琦高若楠叶向彤肖冰洁胡名莉
合成化学 2023年10期
刘天琦, 高若楠, 叶向彤, 黄 益, 肖冰洁, 胡名莉, 何 菱
(四川大学 华西药学院,四川 成都 610041)
螺癸二烯酮作为一种多功能分子骨架,具有广泛的生物活性。螺环化合物的主要特点为2个环共用1个原子,即螺原子(spiroatom)。螺环化合物的刚性非平面结构使其具有良好的稳定性和空间特异性[1],尤其sp3杂化的螺原子赋予螺环分子特殊的立体结构,在生物体内更容易与靶点有效地紧密结合,进而产生影响力大的生物效应[2]。
近10年的研究表明,螺癸二烯酮在抗肿瘤、抗炎、抗病毒以及抗神经退行性等疾病中均表现出良好的生物活性(图1)。其中,螺癸二烯酮的生物活性在抗肿瘤方面的表现尤为突出,如番荔枝素与甜菜碱中均含有螺二烯酮结构,其中番荔枝素具有较高的抗肿瘤活性[3];甜菜碱可以诱导癌细胞的凋亡[4]并可通过抗氧化作用对高血压导致的学习障碍进行保护[5],同时具有肝保护作用[6]。另外,飞龙掌血提取物中的螺二烯酮骨架化合物具有抗流感病毒H2N2的作用[7]。螺杆菌烯A中含有螺环戊烯酮,是一种可用于抗神经炎症和抗神经退行性疾病的药物[8]。此外,合成的三唑螺二烯酮分子对多种癌细胞系均表现出良好的抗增殖活性,可作为抗癌的新型小分子候选药物[9]。
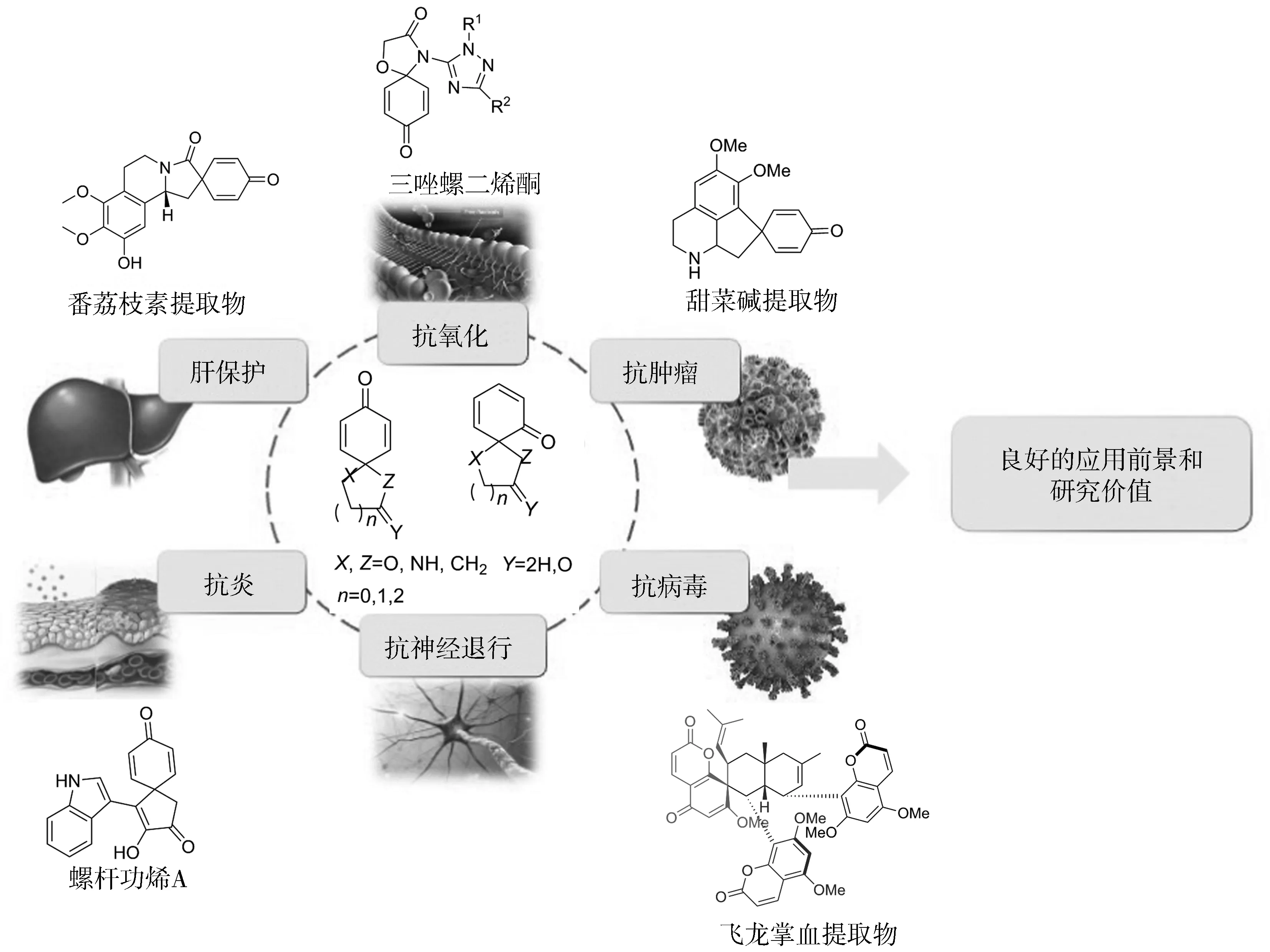
图1 螺癸二烯酮衍生物的生物活性
综上所述,螺癸二烯酮衍生物良好的生物活性使其具有广阔的应用前景和重要的研究价值。基于此,本文将近10年螺癸二烯酮的合成方法进行分类总结。希望能为更多新型螺环衍生物的设计与合成,及其在药物研究领域中的应用提供借鉴作用。
1 通过碳碳键形成构建螺癸二烯酮衍生物
1.1 sp杂化碳参与的分子间反应
(1) 过渡金属催化氧化环化反应
2014年,YUAN课题组[10]首次实现了基于轴向中心手性转移的外消旋联芳基底物的动态动力学不对称转化。在手性Pd-L3配合物催化4-(2-溴芳基)-萘-1-醇(或2’-溴-[1,1’-联苯]-4-醇)与炔的不对称螺环化的反应中,以95%产率,97%对映选择性得到具有手性季碳中心的螺环化合物2(图2)。但该反应区域选择性差,并且底物范围窄,酚类化合物仅局限于萘酚,而未涉及苯酚。2016年,YUAN课题组[11]将两分子的炔烃采用偶联片段进行组合形成双炔类化后物(图3),并使其与简单易得的溴代酚类化合物作用,成功制备了一系列含有螺环骨架的多环分子,解决了上述[2+2+1]螺环化反应的区域选择性问题,实现了卤代苯酚的去芳构化转化。2019年,YUAN课题组[11]直接采用两分子炔烃与简单易得的溴代酚类化合物偶联,省去了双炔底物的合成步骤,通过[2+2+1]反应策略,以高区域选择性实现了萘酚的去芳构螺环化转化(图4)。
为了解决导流装置内腔不易被阳极氧化和电解着色的问题,根据其形状设计了导流装置快速阳极氧化和电解着色专用装置,如图2所示。
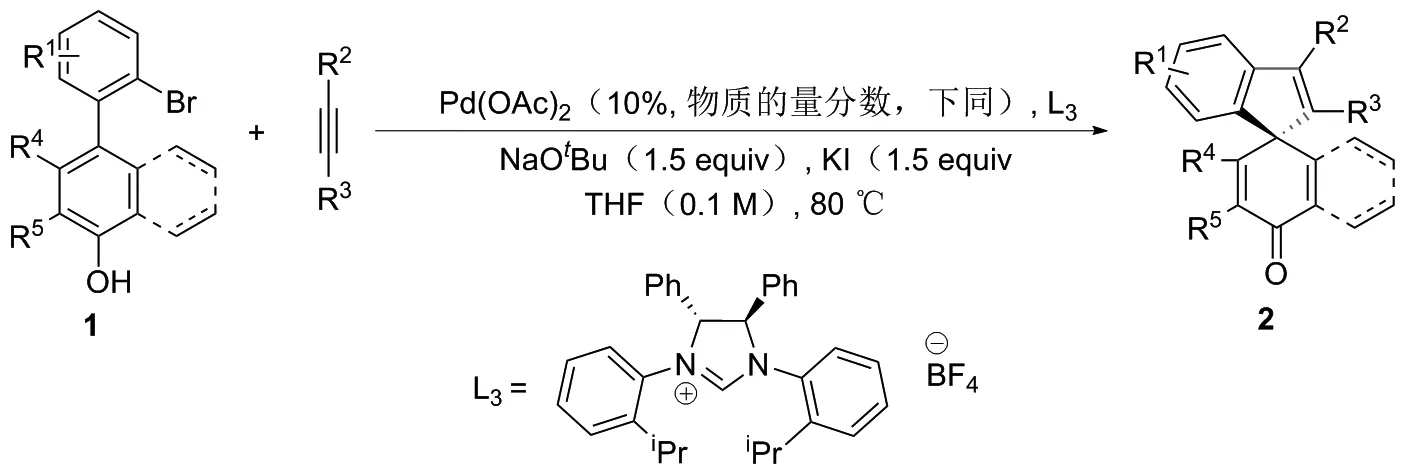
图2 手性Pd-L3催化合成螺癸二烯酮衍生物2的路线

图3 卤代苯酚的[2+2+1]环化反应合成螺癸二烯酮衍生物的路线
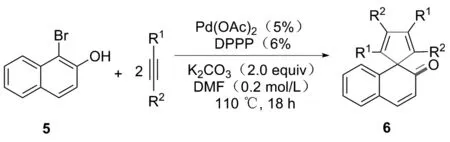
图4 α-卤素-β-萘酚的脱芳[2+2+1]螺环化反应路线
在上述基础上,2015年,ZHENG等[12]通过C(sp2)—H的官能团化/乙炔插入/环化,实现了Rh催化的1-芳基-2-萘酚的不对称去芳构化。在手性Rh催化剂C1、 Cu(OAc)2和空气作为氧化剂的条件下,Rh与羟基络合,C(sp2)—H活化,之后通过炔的插入、脱芳构化和还原消除得到手性螺二烯酮8,产率可达33%~98%,dr最高可达97 ∶3(图5~6)。
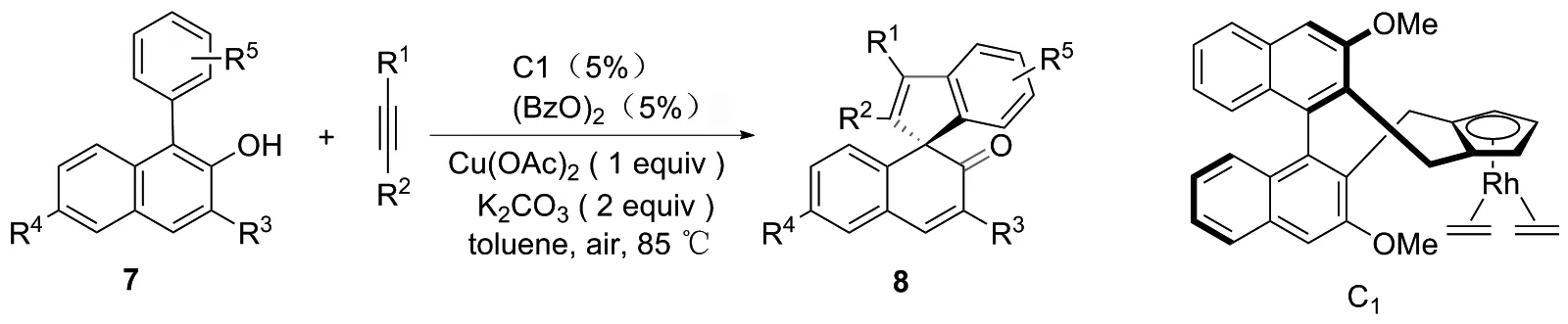
图5 邻-螺烯酮衍生物8的合成路线

图6 手性Cp/Rh催化剂催化合成化合物8的反应机理
上述通过C(sp2)—H的官能团化/乙炔插入/环化,实现了Rh催化的1-芳基-2-萘酚的不对称脱芳构化是利用炔烃在过渡金属催化下发生分子间环加成生成螺环己二烯酮类化合物。该方法是合成螺环己二烯酮的传统方法之一,但存在操作繁琐、原料不易获得、官能团局限和产物收率低等不足。
基于GIS和RDA唐山市降水量空间分布及其影响因素…………………………………………………………邝田萌(4.19)
田间管理的目的在于运用科学、综合的农业技术,为马铃薯植株创造良好的生长发育条件,是促早熟高产高效栽培的重要环节。追肥宜早不宜晚,苗出齐80%后,进行第1次追肥,施碳酸氢铵600~750 kg/hm2(或尿素 225 kg/hm2)左右,追肥后要及时灌水,现蕾期进行培土、浇水。开花初期薯块进入迅速膨大期,结合除草进行第2次培土、浇水,植株封垄前培完土,防止块茎外露变绿,可视植株长势决定第2次追肥,一般不追肥,若需要,可少量追施尿素,约150 kg/hm2。
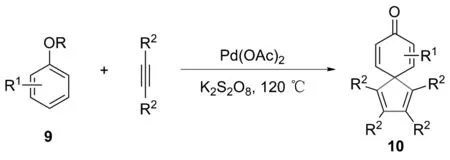
图7 苯酚醚去芳构化制备螺环己二烯酮
(2) 高碘衍生物氧化加成反应
2014年,CHEN等[14]以双三氟乙酰碘苯(PIFA)介导的炔烃加成制备了螺内酰胺衍生物。在PIFA和三氟乙酸(TFA)存在,建立了取代苯甲酰胺和炔烃的无金属参与的氧化环加成反应,在-20 ℃或室温下搅拌反应完成后,加入3 mL饱和NaHCO3溶液,并用二氯甲烷(DCM)提取。合并有机层后再用盐水(20 mL)清洗,用Na2SO4干燥,浓缩,以87%收率获得螺环衍生物12(图8)。
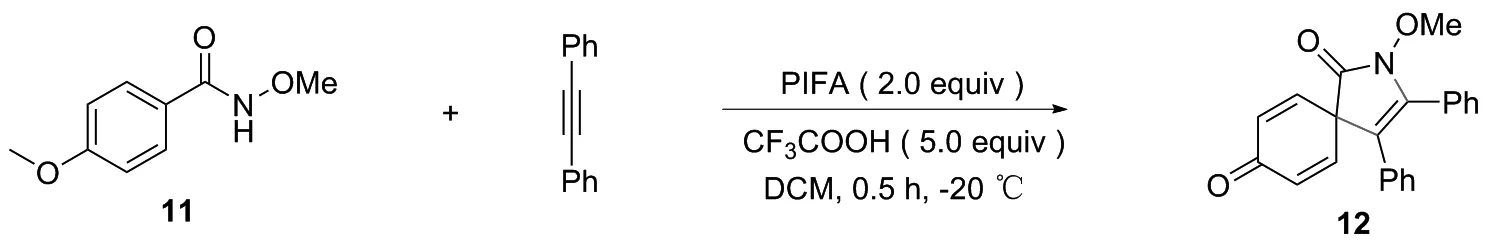
图8 螺环衍生物12的合成路线
(3) 芳基自由基转化成螺二烯酮衍生物
2016年,HEINRICH研究小组[15]发现,2-烯丙基氧基苯基重氮盐13首先放氮产生自由基,继而与炔发生自由基加成形成烯基自由基,随后与2-烯丙基氧基的端基烯发生级联自由基加成后再发生串联自由基攻击芳环,原位环化形成3个C—C键,再经四氢呋喃环开环氧化实现邻螺癸二烯酮的非对映选择性合成,获得邻螺癸二烯酮14。 R2不同,所得产物的产率不同,产率均约为50%(图9)。该实验最关键的步骤为自由基氧化铁(Ⅱ)离子转化成为铁(Ⅲ)离子,并且由实验结果表明,产物14在反应后期阶段的形成速度加快,这不仅说明了铁离子的重要性,而且还表明三价铁作为氧化剂参与了反应。
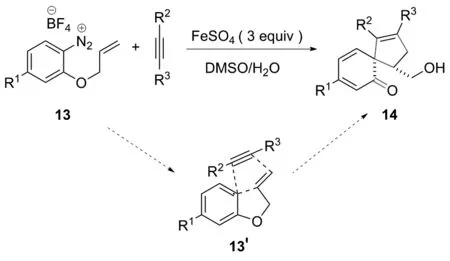
图9 邻螺癸二烯酮14的自由基串联环化反应
1.2 sp杂化碳参与的分子内反应
(1) 过渡金属催化炔衍生物原位环化
金催化在构建复杂分子方面表现出很强的能力。阳离子金配合物可使各种亲核试剂在配位后能够有效地攻击炔烃和烯烃,从而形成螺碳环衍生物。2016年,WU课题组[16]在温和条件下,以1-萘酚衍生物为底物,通过低负载5%催化剂Ph3PAuCl制备了螺癸二烯酮衍生物。该反应对末端炔烃修饰的底物具有良好的反应性。卤素取代炔对金催化的环化没有影响。其他芳香族杂环底物如吲哚、噻吩和吡啶,部分与炔连接,同样可以螺环化。当反应扩大到克级时,仍可达到99%的产率,且反应时间相对较短(2 h),说明该反应的普适性很好。该反应催化循环的路径如下:首先原位生成阳离子金络合物,然后与C≡C键配位并活化。随后发生环化反应,得到烯基金中间体A。最后,中间体A经过质子脱金属化,释放出金催化剂的同时产生所需的螺萘醌产物16(图10~11)。
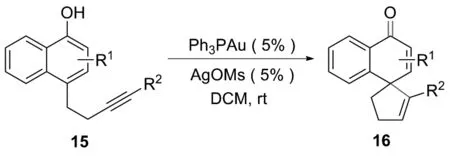
图10 螺环化合物16的合成路线
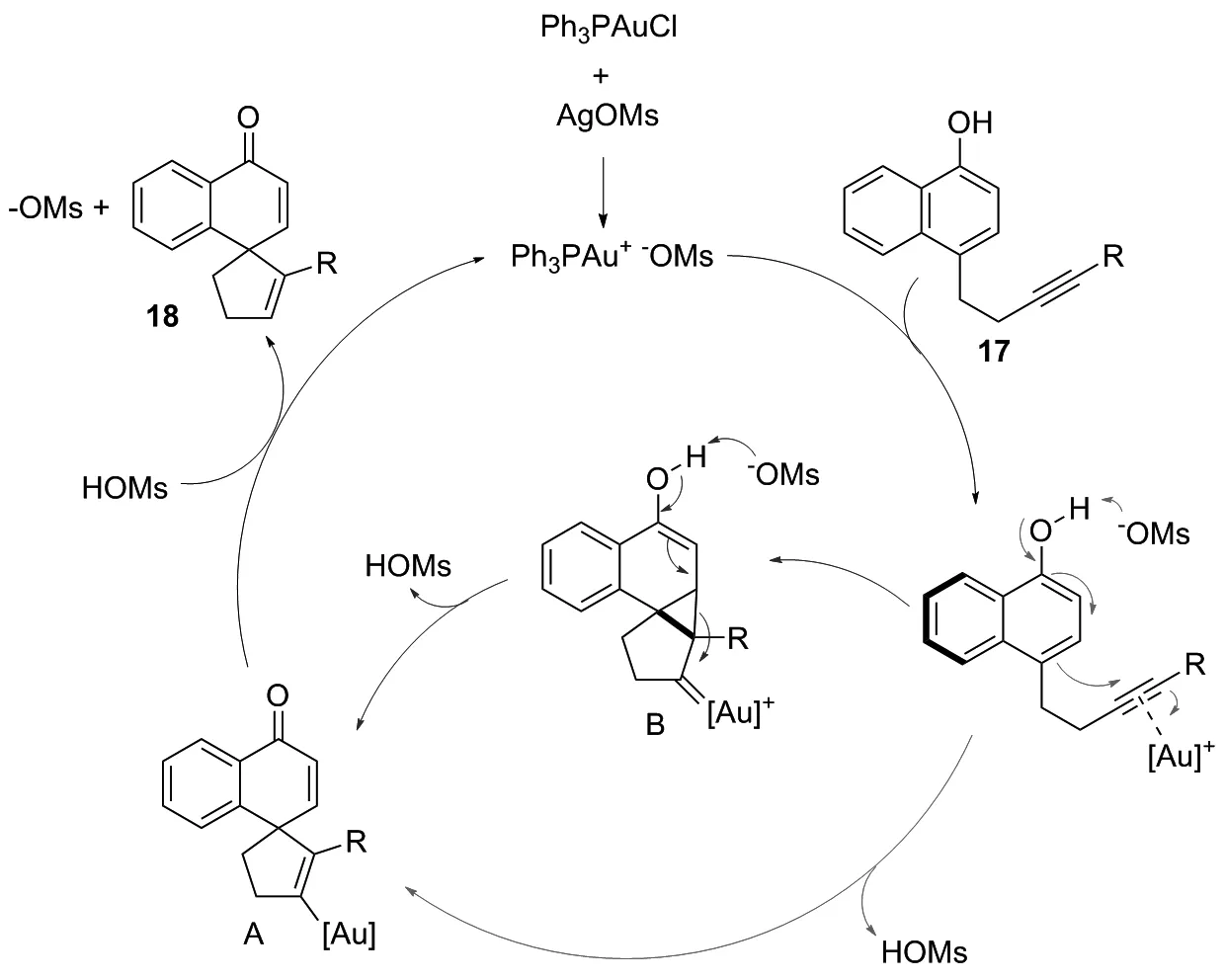
图11 5-内环化(外循环)和1,5-炔环异构化(内循环)合成16的机理
(2) 过渡金属联合有机配体催化苯酚/萘酚衍生物分子内或分子间去芳构化环合反应
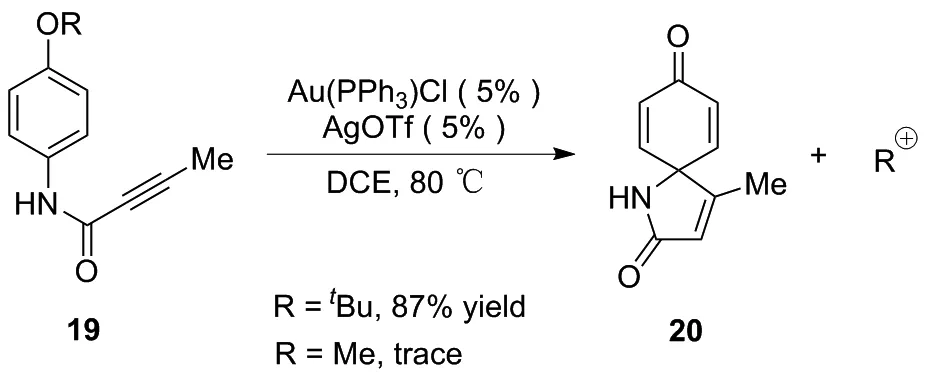
图12 金催化合成氮杂螺癸二烯酮20
观察指标 一般人口学资料(年龄、性别、体质量指数)、吸烟饮酒情况、既往病史(鼻炎、喉炎、支气管炎、肺炎、慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)、高血压、糖尿病、冠心病等)、家族疾病史(高血压家族史、糖尿病家族史、冠心病家族史、乙肝家族史、癌症家族史等)以及哮喘发生结局。
(2) 高价碘催化炔衍生物原位环化
2011年,DOHI课题组[19]报道了一种用活性高价碘剂将炔衍生物原位环化为螺内酯或螺内酰胺的方法,产率可达92%(图14)。该方法以化学计量的双碘代联芳烃C2、m-CPBA、双倍量的对甲苯磺酸一水合物和三氟乙醇为溶剂对取代酰胺进行环化,获得在羰基α位上叠氮基取代的螺内酰胺。类似底物芳基烷基炔衍生物25也可转化为螺环化合物。该反应过程如下:首先,在p-TsOH存在下,用间氯过氧苯甲酸(m-CPBA)原位将C2氧化成高价碘阳离子;然后,底物的炔被亲电碘激活,接着芳环上甲氧基诱导原位环化,形成螺环化碘(Ⅲ)盐。最后,通过形成的盐的还原偶联亲核试剂,从而产生功能化的螺环化合物(图15)。
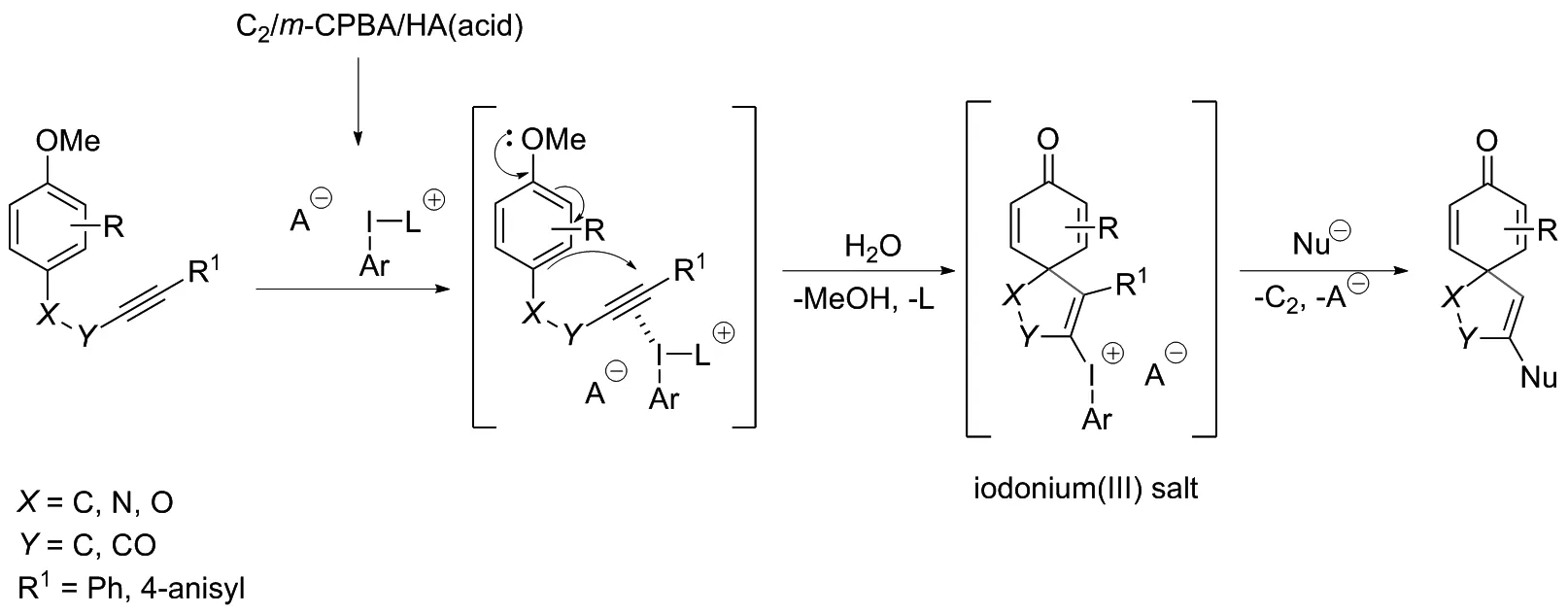
图15 芳基炔碘盐合成螺环的机理
多杀性巴氏杆菌自身不能进行运动也没有芽孢,因此主要是通过患病动物的分泌物和粪便进行传播。首先是感染动物的呼吸道和消化道,出现腹泻等基础性症状;其次也可通过蚊虫的叮咬和血液进行间接传播。其中最简单的传播方式是通过粪便进行传播,家禽家畜、野生动物甚至是人类都可以被传染该种疾病,因此,牦牛一旦出现此类病症,养殖者必须要采取措施进行治疗,以防造成大范围的感染[2]。
As shown in Figure 28, the battery supplies for the motor connected to DC(28 V) port on the right at 0.9 s when two generators and the auxiliary electricity device all fail at 0.8 s. At this situation, only this port can work normally.
研究组出院后的随访依从率达到了93.18%,高于常规组81.82%,差异具有统计学意义(P<0.05)。详情见表3。
(3) 无金属或氧化剂条件下,基于自由基的系列转化反应
(1)N-苄基丙烯酰胺类化合物参与反应
2018年,NECHAEV研究小组[18]以金催化的Ugi环化反应,实现了螺环吡咯烷-2-酮二烯酮衍生物22的非对映选择性合成。该反应为放热反应,通常在数分钟内即可完成,反应条件温和。反应机理如下:首先胺与醛/酮缩合失水转变为亚胺,亚胺被羧酸质子化为亚胺离子,亚胺离子与异腈发生亲核加成生成腈鎓离子,之后羧酸负离子进攻异腈的碳原子生成另一个亚胺中间体,最后亚胺中间体发生Mumm重排反应,发生酰基转移生成Ugi产物(图13)。
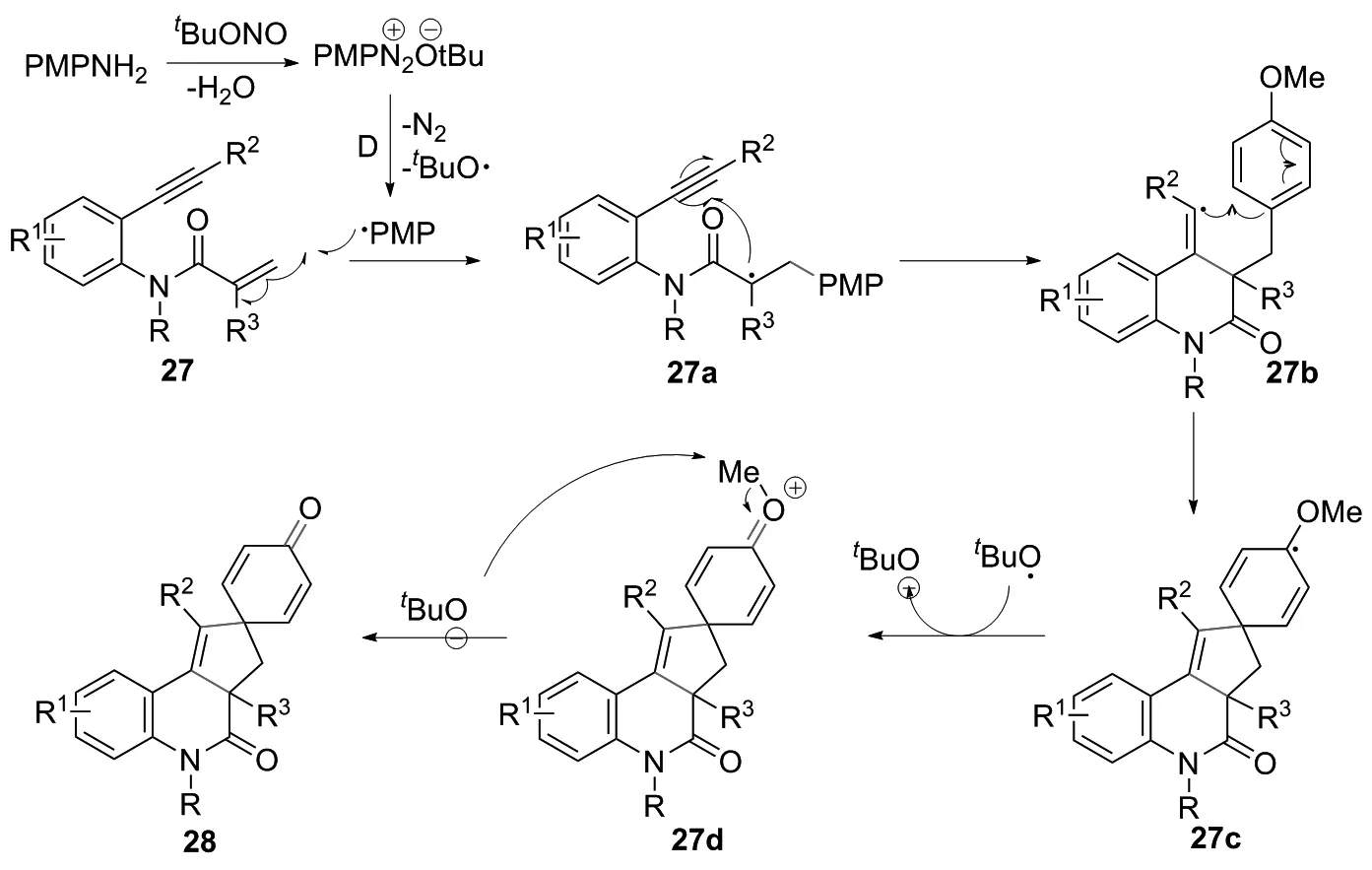
图16 6-exo-dig环化反应或5-exo-trig环化反应合成28的路线及机理
1.3 sp2杂化碳参与的成环反应
2017年,LI课题组[20]建立了1个新的碳中心自由基触发的N-取代1,7-烯炔双环化级联反应,用于合成含螺癸二烯酮的环戊喹啉-4-酮类衍生物。该反应第一步是形成4-甲氧基苯基重氮离子,该重氮离子由苯胺和t-BuONO原位生成,并通过自身分解释放氮气给出PMP自由基和叔丁氧基自由基;所得PMP自由基再加入到末端烯烃中,随后进行6-exo-dig环化反应或5-exo-trig环化反应得到自由基中间体27c。接下来叔丁氧基自由基之间的单电子转移(SET)氧化反应发生,形成中间体27d,其在碱性条件下通过去甲基化最终转化为产物28(图16)。该反应具有条件温和、底物范围广、环化效率高和官能团耐受性高等特点。
氮杂螺环化合物由于其显著的药理活性受到人们的广泛关注,许多重要的药物分子骨架中都含有该类结构。因此,这类分子骨架的合成方法研究将对具有药用价值的分子研究提供方便和借鉴,同时,也可能带来一定的经济效益。反应机理如下:在Ru(bpy)Cl2作催化剂的情况下,N-苄基丙烯酰胺类化合物能与重氮盐在温和的条件下发生光催化反应,有效地形成各种芳基基团取代氮杂螺癸二烯酮化合物。2018年,唐石等[21]以N-(4-甲氧基)苄基烯丙酰胺类化合物29和氟硼酸重氮盐30为反应起始物,Ru(bpy)3Cl2为催化剂,K2CO3为碱,DMF为溶剂,在氮气氛的室温条件下用5 w的蓝色LED灯照射,搅拌反应24 h,得到螺二烯酮衍生物31,产率介于50%~88%之间,反应条件较温和(图17)。其中,R1为2/3-Cl, 2/3-Br, R2为n-Bu,t-Bu,i-Pr, Me, Bn,反应中采用氟硼酸重氮盐(图18)。

图17 可见光下N-苄基丙烯酰胺类化合物去芳构成环反应机理
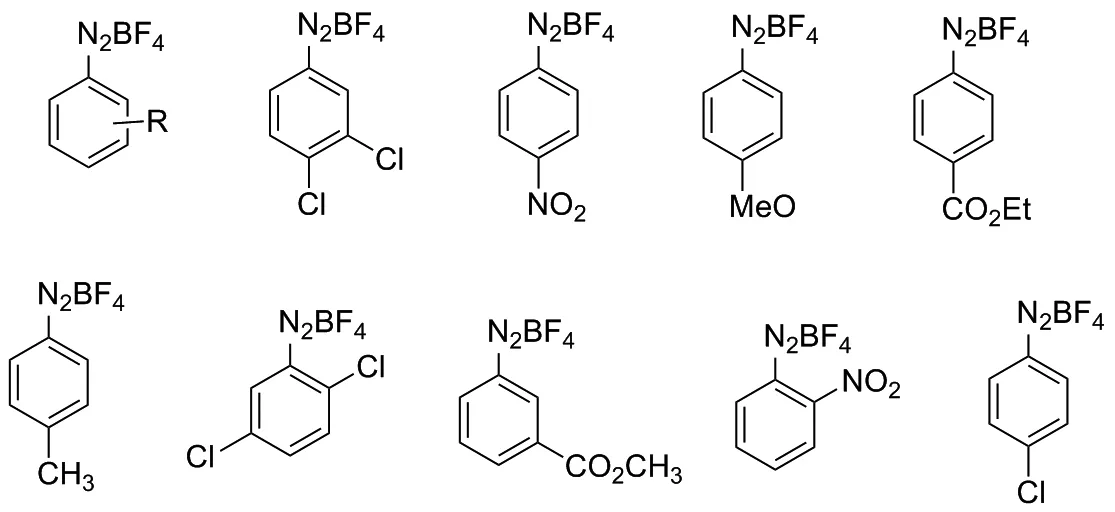
图18 反应中采用的氟硼酸重氮盐
含有卤素的有机分子在生物活性分子中起着重要作用,如渗透性,代谢稳定性等。特别是具有二氯甲基或三氯甲基的多卤代化合物在天然产物中广泛存在,均表现出广泛的生物活性,包括抗肿瘤、抗炎等作用。该类反应的机理是选择性裂解C(sp3)—H键,将多卤代烷烃(例如CH2Cl2和CHCl3)转化为多卤代烷基自由基以引发烯烃的自由基串联环化反应。N-苄基丙烯酰胺类化合物已被证明是良好的自由基受体,容易经自由基介导的串联螺环化反应制备各种不同的氮杂螺癸烯酮类化合物。
进行该类反应的实验操作时应注意以下几点[22]:芳香族四氟硼酸重氮盐为取代或未取代的苯基四氟硼酸重氮盐,取代基优先选择苯基、甲氧基取代的苯基、卤素取代的苯基、甲基取代的苯基、硝基取代的苯基,而最佳选择为苯基四氟硼酸重氮盐、4-甲氧基苯基四氟硼酸重氮盐、4-硝基苯基四氟硼酸重氮盐(排名无先后顺序);碱为无机碱,优先选择碳酸钠、碳酸钾、磷酸钾、碳酸铯和碳酸氢钠中的任意一种,最佳选择为碳酸钠;不使用有机溶剂;N-苄基丙烯酰胺类化合物、多卤代烷烃类化合物、芳香族四氟硼酸重氮盐和碱的最佳投料物质的量之比为1 ∶50 ∶2 ∶2;惰性气体优先选择氩气;最佳搅拌反应时间为12~16 h;后处理和分离纯化得多卤代氮杂螺环己二烯酮类化合物。
2018年,江门市大健康国际创新研究院在克服原有传统制备技术缺陷的基础上,提供了一种苯酚醚去芳香化制备螺环己二烯酮类的方法[13]。该方法将苯酚醚、炔烃、催化剂和氧化剂依次加入到反应管中,从室温升温至120 ℃,反应6~48 h,得到螺环己二烯酮类化合物10,产率为65%~87%(图7)。
此外,该课题组还采用类似的方法设计并合成了一系列磺酰氮杂螺癸二烯酮类新结构化合物,经过初步的体外肿瘤细胞生长抑制实验筛选[33-34],结果发现,化合物60具有较好的抗肿瘤活性,具体合成路线见图29。该方法首先合成N-β-氯乙基取代或未取代芳磺酰胺,之后与苯酚衍生物反应获得苯氧乙基芳磺酰胺。该磺酰胺以高碘化合物为氧化剂,如以碘苯三氟醋酸酯氧化[34]并在醋酸铑存在下得到螺癸二烯酮类衍生物59,经Click反应获得螺环衍生物60。
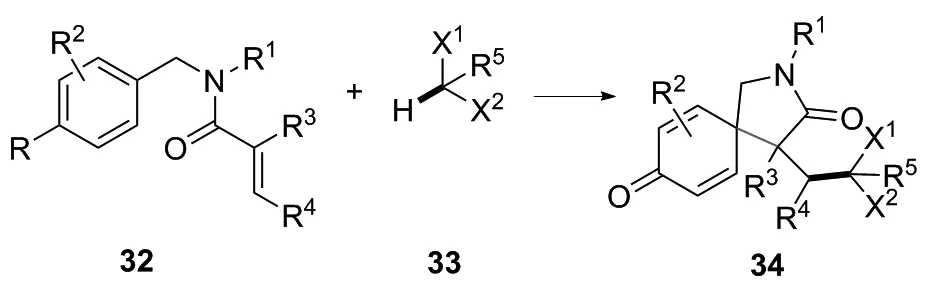
图19 多卤代氮杂螺环己二烯酮类化合物的合成路线
2018年,VACALA课题组[17]发现了一种金催化氧化还原中性的N-芳基烷化剂去烷基化螺环化合成螺内酰胺20的方法(图12)。该反应主要利用C—O键断裂释放稳定的阳离子,并为反应提供驱动力。由于叔丁基阳离子的稳定性高于甲基阳离子,在对-叔丁基氧基底物类似物存在下,螺内酯具有高选择性且产率良好,产率可达87%。而该反应的特点为底物范围广,芳环上可连接供电子和吸电子基团,且酰胺氮也可被取代。芳烃环上对-烷氧基团的共性是通过取代基的差异实现螺环化选择性的关键。虽然对-甲氧基的存在仅会产生极少量螺内酰胺,但对-叔丁氧基底物却能使螺内酰胺产率达87%,表现出高选择性。
2011年,WU课题组[23]完成了以苯酚衍生物为底物的金属及红外光催化的分子内不对称烯丙基脱芳构化构建螺环衍生物。为了克服弱给电子苯环脱芳构化的局限性,可以采用碳酸烯丙基上的丙二酸二酯替代品来稳定苯基碳负离子。由金属铱催化的苯衍生物的分子内不对称脱芳构化,利用铱前体和手性亚磷酸盐配体制备铱配合物,以较高的产量合成了一系列螺[4,5]癸二烯酮化合物36[24]。该法具有良好的对映选择性(99%ee)。该反应中丙二酸酯型取代基的引入是引导苯环亲核反应的关键(图20~21)。
中国城乡二元结构体系的基本特征将公民分为两类,对城镇居民和农民实行不同的政策。然而长期实行这种体制的后果就是农村和城市的发展不均衡,城乡差距越来越大,对农民的生产积极性和生产力造成了严重束缚。打破城乡二元结构体系,可以从户籍制度改革和土地集体所有制入手。政府应采取相关政策,解除户籍制度的限制,实现农民与城镇居民户口的平等;并改革土地所有制,对土地制度进行一定的调整,制定相关详细的法规并加以规范,充分保障农民的土地财产权。只有实行户籍制度和土地制度改革,才能打破城乡二元结构体系,保障农民的土地产权。
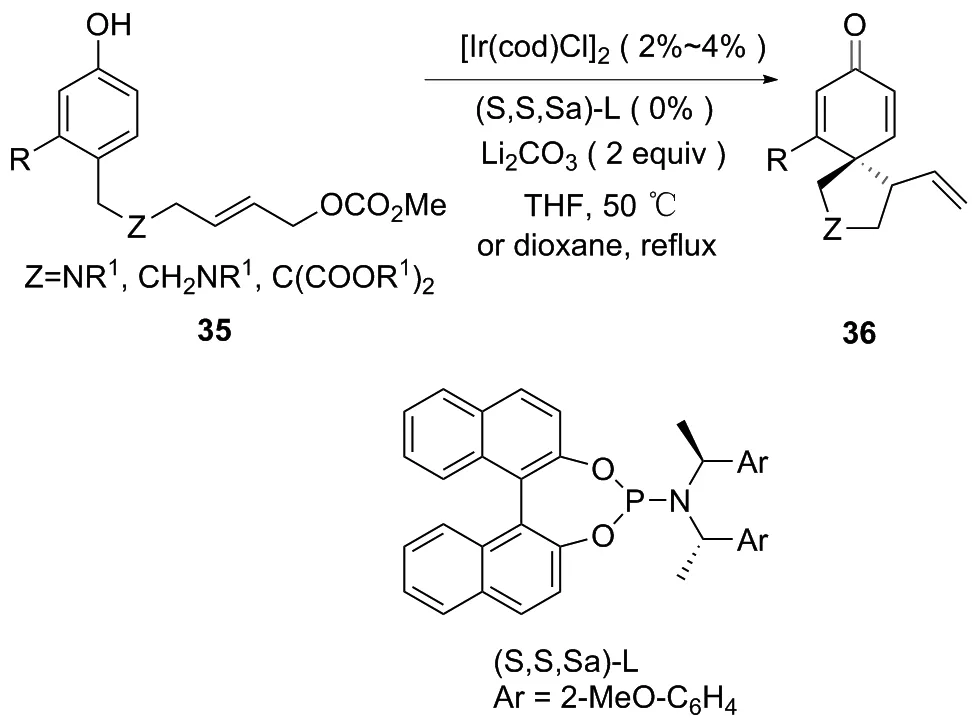
图20 铱催化合成螺环化合物36
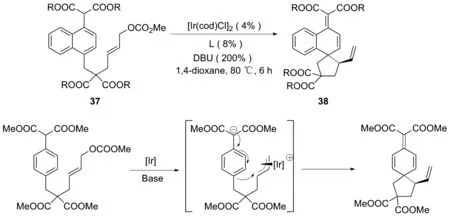
图21 铱催化合成38的路线和分子机制
(3) 金属钯催化的联烯化合物的分子间反应成环
2018年,WU课题组[25]用化合物39合成了由Pd催化的螺[环己烷-1,1’-茚]-2,5-二烯-4-酮。该过程涉及苯酚区域选择性和烯丙基去芳构化,克服了β-H的消除,通过[3+2]环加成有效实现螺环化。本实验的关键步骤为苯酚区域选择性烯丙基脱芳构化,并且通过使用市售的二苯基膦苯基-4-噁唑(PHOX)配体可以实现高对映选择性。其目标分子中末端烯烃,在间氯过氧苯甲酸条件下可被氧化为羰基衍生物40′。通过间氯苯甲酸盐的开环和消除,可进行多元醇的环氧化。在对-甲苯磺酸的条件下,也可异构成环内双键,同时去除TBS保护。当1,4发生加成反应时,还可得到四环产物40″(图22)。
日粮中的蛋白质含量过多或过少均会影响到骨骼发育,然而有学者指出,日粮蛋白水平对体尺的影响不显著[3,12]。邱忠玉等[13]的研究也表明,12%~13%低蛋白日粮对蛋鸡育成期胫长发育无显著影响(P>0.05);郝文博等[14]研究蛋白水平同样对雏鸡胫长无显著影响(P>0.05)。这与本研究结果一致,本研究设定的四种日粮蛋白质水平对京红1号蛋种鸡育成期胫长影响均无显著差异(P>0.05),另外,随日粮蛋白质水平的增加对死淘率无显著差异(P>0.05),表明设定的四种日粮蛋白质水平梯度并未达到能够显著影响胫长和成活率的水平。
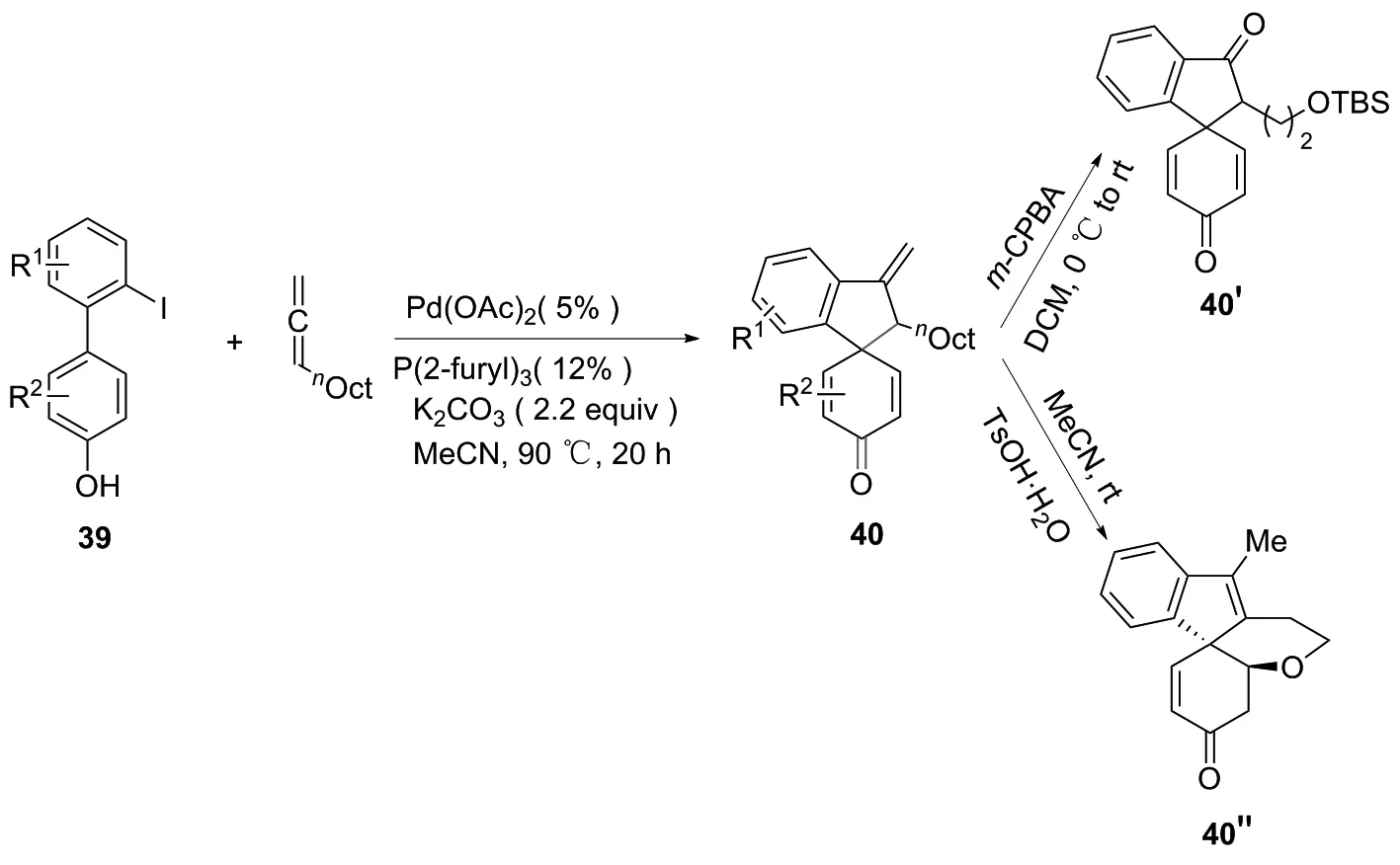
图22 化合物40、 40′和40″的合成路线
(4) 联烯化合物分子内反应成环
ADLER课题组[26]于2015年以α-氨基烯丙基膦酸盐为起始底物,在硝酸铈铵(CAN)处理下合成了螺内酰胺。α-氨基烯丙基膦酸盐不仅在CAN的作用下没有失去对甲氧基苄基(PMP)保护基团,而且是一种很好的N-磺酰基和N-对甲氧基苄基保护剂,并在此条件下还能以80%收率得到螺内酰胺化合物。该实验的关键步骤为亚胺中间体的形成。在第一步中,将对甲氧基苄基氧化成环己二烯酮阳离子41a。接下来,经5-endo-dig环化反应形成亚胺离子41b,41b与水反应形成正离子41c,然后转化为易脱磷酸酯的41d,经脱出亚磷酸二乙酯进而形成内酰胺42。亚磷酸二乙酯可被过量的硝酸铈铵CAN降解(图23)。
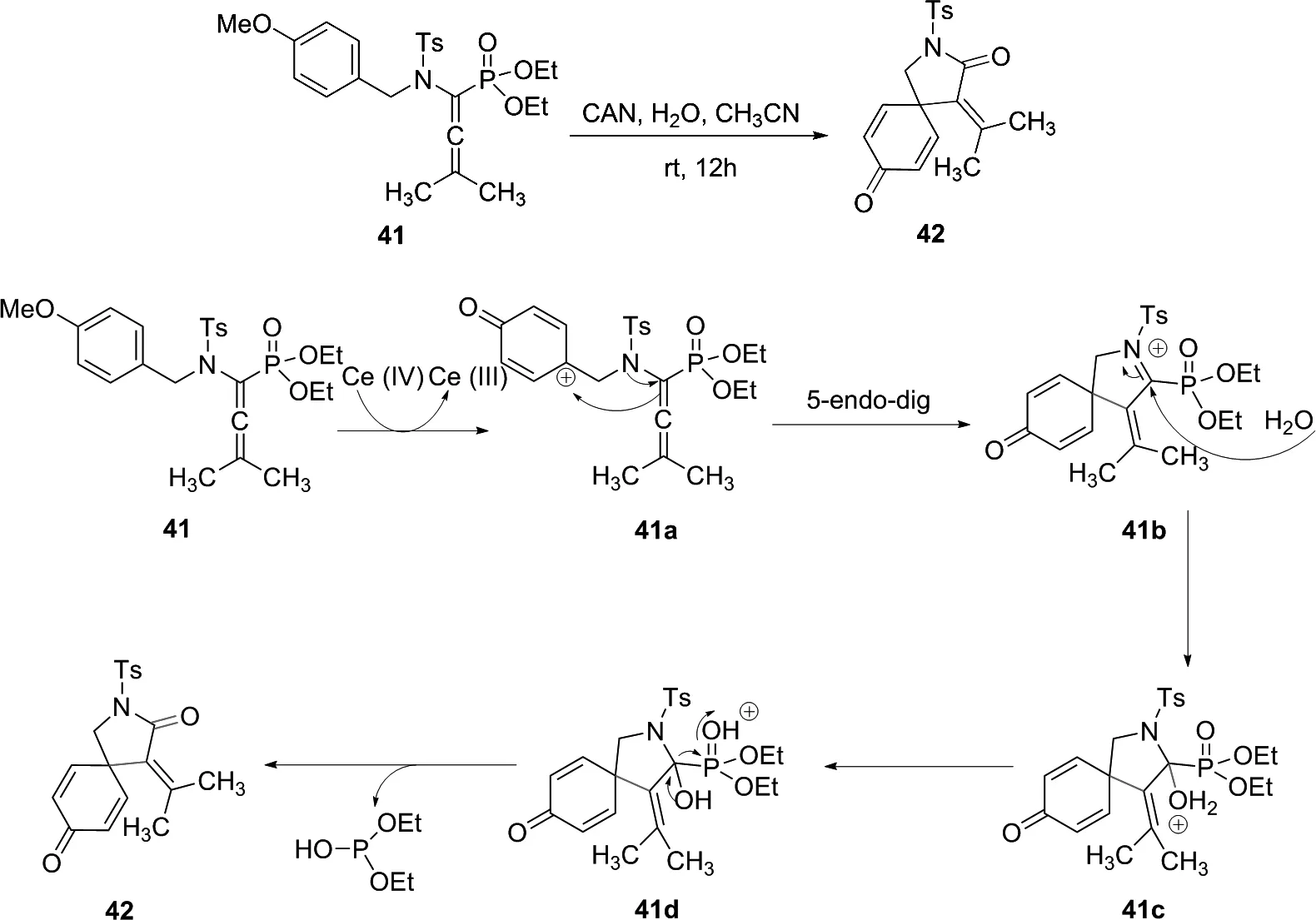
图23 42的合成路线以及硝酸铈铵催化α-氨基烯基膦酸盐合成42的机理
(5) 通过卡宾反应成环
2017年,NAKAYAMA课题组[27]以(S)-TRIPAg作为手性催化剂成功合成了螺癸二烯酮衍生物,避免了C—H插入或布赫纳反应。该研究实现了银卡宾介导的化学和高度对映体选择性苯酚去芳构化,底物普适性好。化合物43与(S)-TRIPAg反应形成有机银配合物。经脱芳构化和脱金属后,得到了含有螺癸二烯酮支架的化合物45(图24)。

图24 羰基银合成45的路线及机理
1.4 sp3杂化碳参与的成环反应
2018年,TAN课题组[28]首次将C(sp3)—H的活化和萘酚去芳构化结合起来,开发了螺环骨架的形成。化合物46经过C(sp3)—H活化过程形成关键中间体(限速步骤)。然后,与萘酚47交叉耦联脱芳构化,以83%产率得到邻螺癸二烯酮48(图25)。
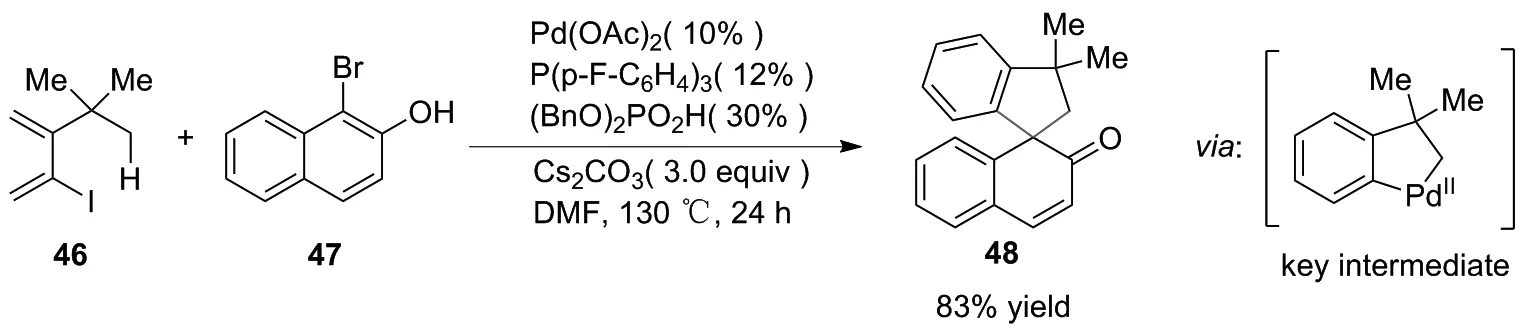
图25 采用C(sp3)—H活化/萘酚去芳构化级联反应合成48的路线
2 通过碳-氮键形成构建螺癸二烯酮衍生物
2.1 酰胺氮为氮源
2012年,何菱课题组[29]首先以醋酸铑为催化剂,有效催化对位无取代基的芳香醚49,经氧化环合成螺癸二烯酮衍生物50(图26)。合成机理如下(图27):首先,在二乙酰氧基碘苯和铑盐的存在下与4-甲基-N-(2-苯氧乙基)苯磺酰胺中的氮形成铑配合物。这种酰亚胺-铑配合物可能与PhI(OAc)2中的碘偶联,从而在苯环的1位和4位同时形成C—N和C—Rh键。其次,PhI(OAc)2中的1个OAc基团亲核进攻形成中间体B。随后,中间体B经分子间氢转移形成阳离子III,释放PhI和HOAc。最后,由于化合物A的不稳定性,在溶液中发生消除得到产品50,产率70%。本反应的优势首先为反应条件温和,室温下即可反应且底物范围广;其次,该反应采用了一种高区域选择性的一锅反应方法;此外,该反应中使用的催化剂较简单。
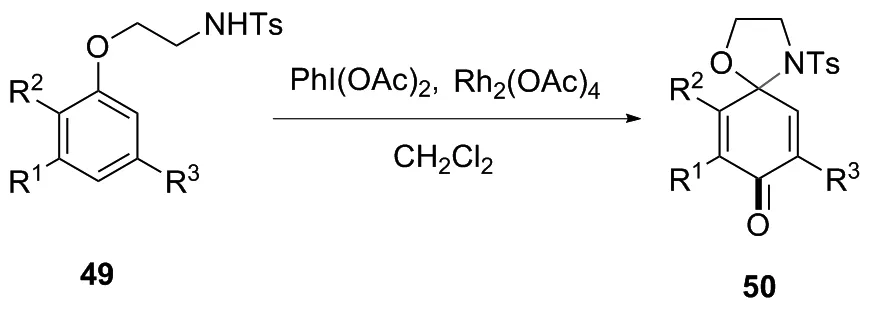
图26 螺二烯酮衍生物的合成路线
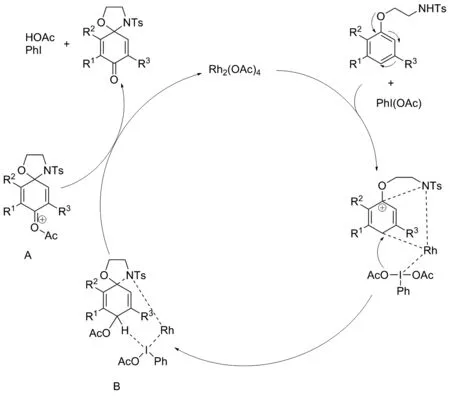
图27 催化氧化合成50可能的反应机理
2.2 磺酰胺氮为氮源
醌型结构单元的衍生物多具有抗肿瘤活性[30]。近年来也有大量磺酰胺类化合物作用于不同的靶点并显示出高度的选择性和特异性,其作用机制是多样的,如干扰微管蛋白聚合、阻滞细胞周期正常进行、抑制碳酸酐酶、叶酸依赖性酶、甲硫氨酰氨肽酶和组蛋白去乙酰酶、抑制血管内皮细胞生长因子等[31]。磺胺类化合物结构中的芳磺酰胺具有浓聚于肿瘤的特性[31],具有潜在的肿瘤靶向性。因此,何菱课题组将磺酰胺与醌型结构单元结合于一体,在具有抗肿瘤活性的氮杂螺癸二烯酮衍生物的基础上引入磺酰胺基团,为高效低毒的抗肿瘤新药开发奠定基础[32](图28)。
图19中R表示OR’, R’选自C1-12烷基、C6-20芳基、C1-12烷基、三(C1-12烷基)硅基、C1-12酰基;R1选自C1-12烷基、C6-20芳基-C1-12烷基、C3-8环烷基;优先选择C1-8烷基、C6-20芳基-C1-6烷基、C3-6环烷基;R2选自氢、卤素、C1-6烷基、C1-6烷氧基;R3、 R4选自氢、C1-6烷基;R5选自氢、C1-6烷基、C1-6卤代烷基;X1、 X2表示卤素。本合成方法具有宽泛的反应底物适应范围,目标产物分离产率高达91%。

图29 化合物60的合成路线
2.3 多氮杂环胺为氮源
2017年,何菱课题组[35]以Cu2+催化氧化胺化,实现三唑联螺癸二烯酮衍生物的合成,同时,类似方法也可合成新型三氮唑联螺二烯酮苯并衍生物[29]。该类合成均可按图30所示合成。首先,用高效的一锅法对醛(61)的氧化氰胺化,制备取代N-氰基亚胺酯63。然后,对相应的N-氰基亚胺酯进行环化反应,形成1,2,4-三唑衍生物64[36]。其次,将酸62与二氯亚砜反应转化为酰氯,1,2,4-三唑衍生物64的氨基与酰氯反应得N-(1,3-二取代基-1H-1,2,4-三唑-5-烷基)-2-苯氧乙酰胺65,65经还原得到1,3-二取代基-N-(2-苯氧乙基)-1H-1,2,4-三唑-5-胺67。随后以PhI(OAc)2为氧化剂,Cu(CF3SO3)2为催化剂,通过氧化胺化反应形成目标衍生物66和68。
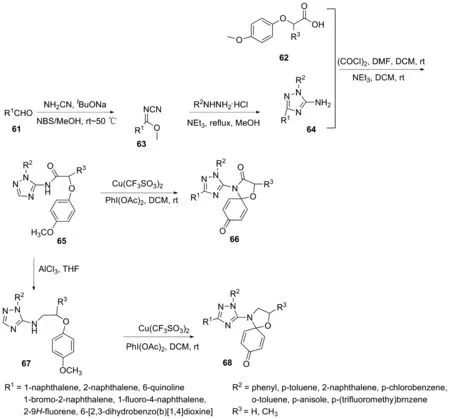
图30 化合物66和68的合成路线
2018中国游戏产业年会中,中宣部出版局副局长冯士新表示,“首批送审游戏”已经完成审核,正在抓紧核发版号,游戏股随即出现大涨。2015年至今,行业的增速开始放缓,而游戏股股价可谓腰斩再腰斩,如今游戏版号有望松绑,行业能否再次迎来爆发,个股是否存在投资机会?
学音乐的都知道,该高调的时候就要高调,如果该高调的时候低调了,那叫跑调。——周立波告诫,不要相信“做人要低调”这句话
此外,2020年,SAHOO和SARKA[37]报道了N-取代螺氮杂环二烯酮、二氢螺环-5氮杂环二烯酮和螺内酰胺的立体选择性合成方法(图31~32)。当其以四氢呋喃为溶剂时,可获得氮杂螺二烯酮70a和氮杂二氢螺二烯酮70b。随着PTAB[苯基(三甲基)三溴化铵]与水的当量增加,70b产率提高。当PTAB(1.5 equiv)和H2O(2.0 equiv)加入时,产物70b产率可达78%~80%。当其以甲醇为溶剂时,主要获得螺内酰胺70c产物。反应机理为:在碱性条件下取代萘酚形成萘氧离子,然后萘氧离子与三溴铵盐相互作用,使其δ-氮有利于亲核进攻形成反应中间体A(图32)继而发生分子内关环生成所需的邻螺氮杂环[38]。
导师对研究生的培养倾向于选择与其科研项目相关的知识、方法和目标,缺乏对研究生进行系统、全面的专业知识、科学方法、科研能力和学术道德的培养。有调查发现,52%的研究生认为“参与导师的科研项目对自己的科研能力提升程度”有“部分帮助”或者“很少帮助”;73%的研究生认为“导师课题和自己研究兴趣的一致程度”“部分一致”或“不一致”。[2]87
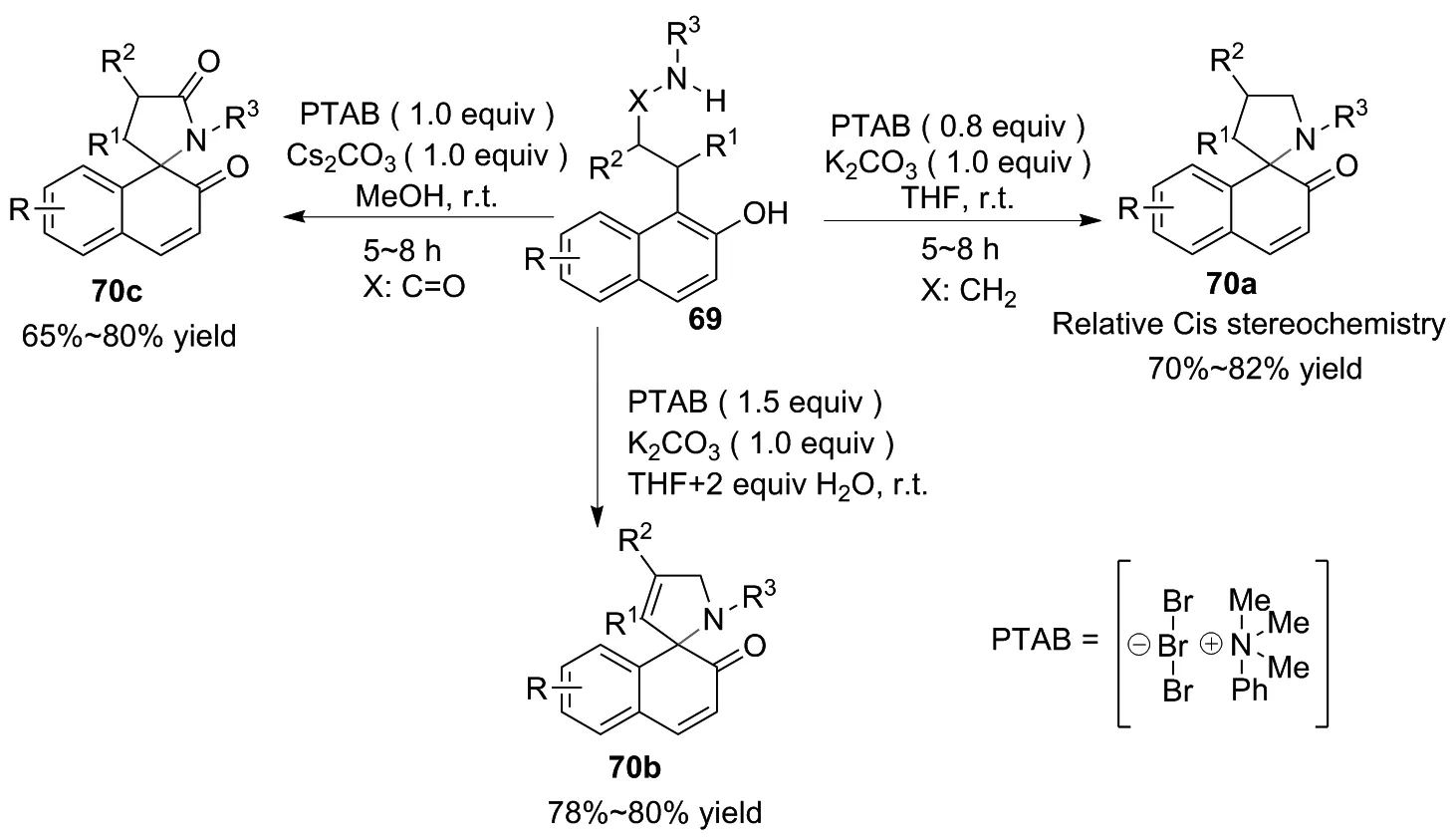
图31 化合物70的合成路线

图32 PTAB参与的70的合成机制
3 通过碳-氧键形成构建螺癸二烯酮衍生物
3.1 羧基氧作为氧源成环
(1) 碘试剂催化苯酚衍生物的环化反应
2008年,DOHI课题组[39]发现一种新刚性手性螺环骨架的碘(III)试剂(R)-C1a,并对1-萘酚衍生物71进行对映选择性氧化,获得手性螺内酯72(图33),产率66%~86%,ee78%~86%。而(R)-C1a的获得可在m-CPBA存在下,由(R)-C1b原位生成,同样可通过上述反应得到相应的螺环产物72[40]。 2013年,DOHI团队对刚性手性螺环胺骨架碘(III)试剂进行改性,在碘原子邻位引入乙基,得到(R)-C1c。该催化剂提高了反应的收率和对映选择性,并成为最佳催化剂[41-45]。该反应主要是以酚氧原子与碘(III)中心形成A,以利于亲核试剂进攻中间体A,得到氧化产物。另外,碘苯从中间体A中释放后,形成邻苯醌阳离子B,紧接着被亲核试剂进攻,生成螺环产物。

图33 碘(III)催化合成邻螺醌衍生物72的路线
2015年,莫托和同事为了合成螺内酯蒿素74[46],将蒿素73在LiOH水溶液中水解,水解产物以四氢呋喃-甲醇-水(3 ∶1 ∶1,V∶V∶V)为溶剂在50 ℃条件下反应2 h进行去芳香化,再与氧化剂PIFA在22 ℃条件下反应12 h。最后柱层析分离纯化螺环产物74,产率56%(图34)。反应过程可能存在2种可能的中间体:73与PIFA交换形成中间体A或解离过程中的阳离子中间体B。这种方法有助于将功能化的苯并香豆素转化为螺内酯。
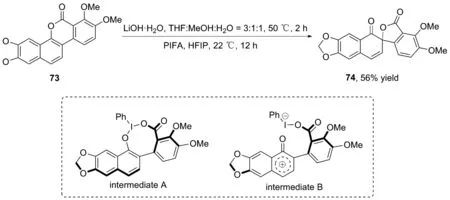
图34 化合物74的合成路线
(2) 光催化非酚类联芳烃的环合反应
2019年,WANG等[47]首次报道了羧基介导的非酚类联芳烃光催化去芳构化反应。底物75在条件I(无水无氧、室温、与TEMPO、 DABCO反应4 h)或条件II(有水有氧、室温、与DDQ和H2O反应15 h)下反应。在这2种不同体系中反应,大多数底物均可转化为螺内酯,获得中高产量。其反应机理如下:第一步均为关键中间体A羧基自由基的形成。第二步,羧基自由基中间体A通过分子内环化促进去芳构反应生成中间体B。第三步,分别在I、II两条件下进行。在I条件下,中间产物B被氧亲核进攻生成过氧自由基C,C与B相互作用生成自由基E,E在TEMPO存在下通过氢原子转移进一步
猜你喜欢
食品安全导刊·中旬刊(2022年3期)2022-04-15
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
世界农药(2019年3期)2019-09-10
中学生数理化·高二版(2016年3期)2016-12-26
中国粮油学报(2016年1期)2016-02-06
合成化学(2015年10期)2016-01-17
化工进展(2015年3期)2015-11-11
核科学与工程(2015年2期)2015-09-26
中南民族大学学报(自然科学版)(2014年4期)2014-08-06
