
Strong anharmonicity-assisted low lattice thermal conductivities and high thermoelectric performance in double-anion Mo2AB2(A=S,Se,Te;B=Cl,Br,I)semiconductors
2023-11-02 08:12HaijunLiao廖海俊LeHuang黄乐XingXie谢兴HuafengDong董华锋FugenWu吴福根ZhipengSun孙志鹏andJingboLi李京波
Chinese Physics B 2023年10期
关键词:原文
Haijun Liao(廖海俊), Le Huang(黄乐),2,†, Xing Xie(谢兴), Huafeng Dong(董华锋),Fugen Wu(吴福根), Zhipeng Sun(孙志鹏),‡, and Jingbo Li(李京波)
1School of Materials and Energy,Guangdong University of Technology,Guangzhou 510006,China
2Guangdong Provincial Key Laboratory of Information Photonics Technology,Guangdong University of Technology,Guangzhou 510006,China
3School of Physics and Optoelectronic Engineering,Guangdong University of Technology,Guangzhou 510006,China
4School of Physics and Electronics,Hunan Key Laboratory for Super-microstructure and Ultrafast Process,Central South University,Changsha 410083,China
5College of Optical Science and Engineering,Zhejiang University,Hangzhou 310027,China
Keywords: thermoelectricity,anharmonicity,lattice thermal conductivity,anisotropy,first-principles calculations
1.Introduction
For the past few years,the problem of the fossil fuel crisis and climatic issues has become more and more serious,and thermoelectricity(TE)materials have been widely studied due to their low-pollution-environmental-friendly application.[1-5]The energy conversion efficiency in TE technology is mainly by the dimensionless figure of merit of the employed material.It is calculated asZT=S2σT/κ, whereSis the Seebeck coefficient,σis the electric conductivity,κis the thermal conductivity including charge carrier(κe)and lattice(κl)contributions,Tis the absolute temperature.[6]Several excellent reviews describe the rules for enhancing TE efficiency by energy-band engineering,[7-10]interfacial engineering,[11]impurity doping,[12-14]mosaic microstructure,[15]and component alloying.[16-18]
The key difficulty in obtaining high TE property with highσand lowκlies in reduce the correlation between electron transport and phonon scattering, which is usually hard to carry out.From the definition of the thermoelectricity figure of merit, it is clear that material with high electric conductivity and Seebeck coefficient or low thermal conductivity can actually be an ideal TE material.Whereas, using a straightforward way to get high TE performance is a difficult work because of these strong coupling of properties above.It is demonstrated that some special and complex crystal structures will reduce thermal conduction without decreasing electronic transport.[19,20]The two-dimensional(2D)SnSe can getZT ≈4.31 at 500 K, through properly adjusting interlayer distance to restrain thermal transport and to improve electric transport.[21]Excellent TE properties were achieved in doped clathrate compounds, in whichσandκare weakly correlated due to the weak coupling between guest atoms and the clathrate framework.[22]Theoretical and experimental works demonstrated that weak coupling betweenσandκrequires low-frequency phonon vibrations,resulting in amplified asymmetric atomic vibrations and enhanced anharmonicities.This strategy was employed in many semiconductors,such asα-MgAgSb[23]and Cu3SbSe3[24]to obtain high TE performance.
It is established that low-frequency phonon vibrations with strong anharmonicities usually appear in TE materials with complex structures which usually exhibit bonding heterogeneity transport anisotropy.[25,26]Double-anion semiconductors have been proved to be promising TE materials with complex structures.As a typical double-one, Bi2O2Se, actually achieves higher carrier transport and lower thermal conductivity compared to Bi2Se3.[27,28]Al2A2Se2(A=Cl,Br,I)exhibit anisotropic thermal conductivities due to the directiondependent Al-Se bonding.[29]Monolayer In2Cl2O2[30]and Ga2I2S2[31]show strong anisotropies in their power factors and low-level thermal conductivity.
As another class of double-anion semiconductors, transition metal sulfide halides,TM2AB2(TM=Mo, W;A=S,Se, Te;B=Cl, Br, I) have been explored to have excellent anisotropic electric transport properties.[32]The rather complex crystal structures and low lattice symmetry of these materials make them promising TE semiconductors.Further studies to explore the TE properties ofTM2AB2and the underlying mechanism are in much desire.The unique natures with respect to other aforementioned double-anion materials in the TE properties ofTM2AB2are also far from explored.
In this context, we studied the structural and electronic properties of layered Mo2AB2(A= S, Se, Te;B= Cl, Br,I) materials to explore their promising TE performance by first-principles approach.Excellent TE properties with highσand lowκare revealed.The lowκis attributed to soft phonon modes with strong anharmonicity.Direct Mo-Mo couplings further reduce the lattice thermal conductivities and increase the anharmonicities by introducing low-frequency phonon modes.TE properties of these Mo2AB2monolayers exhibit obvious anisotropies due to the direction-dependent chemical bondings and transport properties.The double anions with distinct electronegativities of Mo2AB2monolayers benefit much to their good TE properties.These results provide a theoretical understanding on the TE properties of double-anion compounds, and provide instruction to realize high TE performance in Mo2AB2semiconductors by selecting the type of anions and regulating carrier type and concentration.
2.Methodology
Accurate electronic structures are calculated by using hybrid Heyd-Scuseria-Ernzerhof (HSE06) functional with the default mixing parameter of 0.20, as implemented in PWmat package.[38-40]The phonon dispersion of all the employed systematically calculated by density functional perturbation theory method[41]with the second-order interatomic force constants (IFCs) considered, as implemented in PHONOPY software.[42,43]The third-order IFCs are obtained by resolving the irreducible set of atomic displacements with assist of the THIRDORDER.PY script.[44]A 2×1×1 supercell with thek-grid mesh of 2×2×1 is used in the second-and thirdorder IFCs calculations with the energy and force convergence criteria of 10-8eV and 10-6eV/°A,respectively.The cut-off radius of the nearest interaction is set as 1.40 nm in the thirdorder IFCs calculations.All the parameters related to the thirdorder IFCs calculations are tested for convergence, as shown in Fig.S3 in supporting information.
Based on the second- and third-order interatomic force constants, the temperature-dependent lattice thermal conductivities (κl) are obtained by ShengBTE software package[44]through solving the linearized phonon Boltzmann transport equation(BTE)
wherekBstands for the Boltzmann constant,andTis absolute temperature.¯his the reduced Planck constant.f0stands for the Bose-Einstein distribution.Ωis defined as the volume of the unit cell andNis the number of Gamma-centeredk-points,N=10×10×1 in first BZ for this work.Theωλandυλare the angular frequency and phonon group velocity of the phonon modeλ, respectively.The component of the phonon mean free path (MFP) along thebdirection is represented as.
The electric transport properties as functions of carrier concentration and temperature are calculated using BOLTZTRAP code.[45]The electric conductivityσand Seebeck coefficientSare expressed as
wheref0represents the equilibrium Fermi distribution function.ηis the chemical potential.τkandυkstand for the carrier relaxation time and group velocity of an electron at thekstate, respectively.The deformation potential (DP) theory is employed to calculate the carrier mobility and carrier relaxation time, and the details of these calculations are given in supporting information.
3.Results and discussion
Bulk Mo2AB2(A=S,Se,Te;B=Cl,Br,I)holds space group ofPmmawith layered structure.Figure 1(a)shows the crystal structure and the first BZ of monolayer Mo2AB2.The unitcell of monolayer Mo2AB2contains total 30 atoms due to its relative low symmetry in comparison with transition metal dichalcogenides.The in-plane lattice constants of monolayer Mo2AB2are listed in Table S1.Due to the orthogonal crystal structures, these monolayers present obvious anisotropic features in their structural and transport properties.[32]

Fig.1.(a) Crystal structures of Mo2AB2 monolayer.The interlayer direction is redefined to along the c direction.Its first Brillouin zone with high-symmetry points labeled is also plotted here.(b)Calculated Young’s modulus (blue and orange points), Y, and bulk modulus (red points),K,of MoAB2 monolayers.
To evaluate the thermodynamic stability of these monolayers, the phonon dispersions are calculated in Fig.2.It is found that monolayer Mo2SB2(B= Cl, Br, I), Mo2SeCl2,and Mo2SeBr2show no imaginary frequencies in their phonon spectra,indicating they are dynamically stable.While phonon dispersions of other four Mo2AB2monolayers employed in this work present an imaginary frequency at S (0.5 0.0 0.0)point.It is found that heavierAandBanions tend to reduce the dynamic stability of Mo2AB2monolayer.Additionally,for a given vibration mode,its phonon frequency decreases as the atomic masses ofAandBincreases.Consequently,the speed of sound also decreases with the atomic number ofAandBions, following the inverse proportionality between the speed of sound and the atomic mass.
Exploring the mechanical properties of a material is usually helpful to evaluate its thermal transport properties.In Fig.1(b) and Table S1, elastic constants,C11,C22,C12,C66and associated Young’s modulusY, and bulk modulusK, are calculated.The elastic constants of all the structures satisfy the thermostable rules in orthogonal symmetry(C11>0&C22>0&C66>0&C11·C22>C212).Our results indicate that all the Mo2AB2monolayers exhibit good mechanical stability with moderateYandKvalues.[32]High anisotropies are observed in their Young’s modulus due to their anisotropic characters of chemical bondings.Especially,as the atomic sizes ofAandBatoms increase, the anisotropies of mechanical properties are enhanced.Detailed analysis of their crystal structures can provide clear evidence for the increasing anisotropy.Specifically,it is observed that the Mo-Abondings exhibit high-covalency characters along thebdirection, which results in the larger Young’s modulus along this direction than along theadirection.As a result,the acoustic velocity and associated thermal conductivity along thebdirection is expected to be higher than theadirection.Furthermore, it is noted that these Mo2AB2monolayers have low bulk modulus about 50 N/m.So relative weak chemical bonding is expected in Mo2AB2monolayers.It can be assumed that these Mo2AB2monolayers should exhibit low thermal conductivities due to their soft lattices with high ionic bondings.
原文:You’re not the sharpest knife in the drawer,Pops.That’s his music.
From the phonon dispersions in Fig.2, we also found that acoustic velocities corresponding to transverse acoustic(TA), longitudinal acoustic (LA) and the third acoustic (ZA)modes are different from each other.It should be mentioned that all these three acoustic modes exhibit weak dispersions in the image, resulting in low acoustic velocities.Furthermore, there is no energy gap between acoustic modes and low-frequency optical phonon modes,indicating the existence of strong phonon scattering between low-frequency optical modes and acoustic modes.These results reveal that there exists relative strong three-phonon scattering in Mo2AB2monolayers.Consequently, lattice thermal conductivity can be restricted by this three-phonon scattering restrains the thermal conductivity, severely enhancing the thermoelectric performance of these materials.
Electronic properties and orbital coupling characters are intimately related to the thermoelectric properties of a material.From the electronic band structures in Fig.S1, it is found that all Mo2AB2monolayers have direct bandgaps at the S (0.50.00.0) point of the first BZ (Fig.1(a)).Accurate electronic structures are obtained by using HSE06 functional method.As shown in Fig.3(a),the bandgap of Mo2AB2monolayer shows significant dependence on the atomic number ofAand weak dependence on that ofBanions.Further evidence can be obtained from their projected density of states(PDOS)in Fig.S2.Compared to chalcogenideAions,Bions have major contributions to conduction band states.The VBM states are mainly composed of Mo 4d andAp orbitals.Additionally,Mo2AB2monolayers are expected to have similar magnitude of Seebeck coefficients due to their rather similar general features in their electronic structures near the Fermi level.In Fig.3(a), energy positions of bandedge states, the VBM and CBM states,of these Mo2AB2monolayers with respect to the vacuum level are calculated on the HSE06 level.For givenAin Mo2AB2, energy positions of the VBM and CBM increase with the atomic number ofB.As the atomic number ofAincreases, the VBM states shift up, but inverse trend for CBM state.This is mainly attributed to the increasing atomic level ofB(orA)p orbitals.
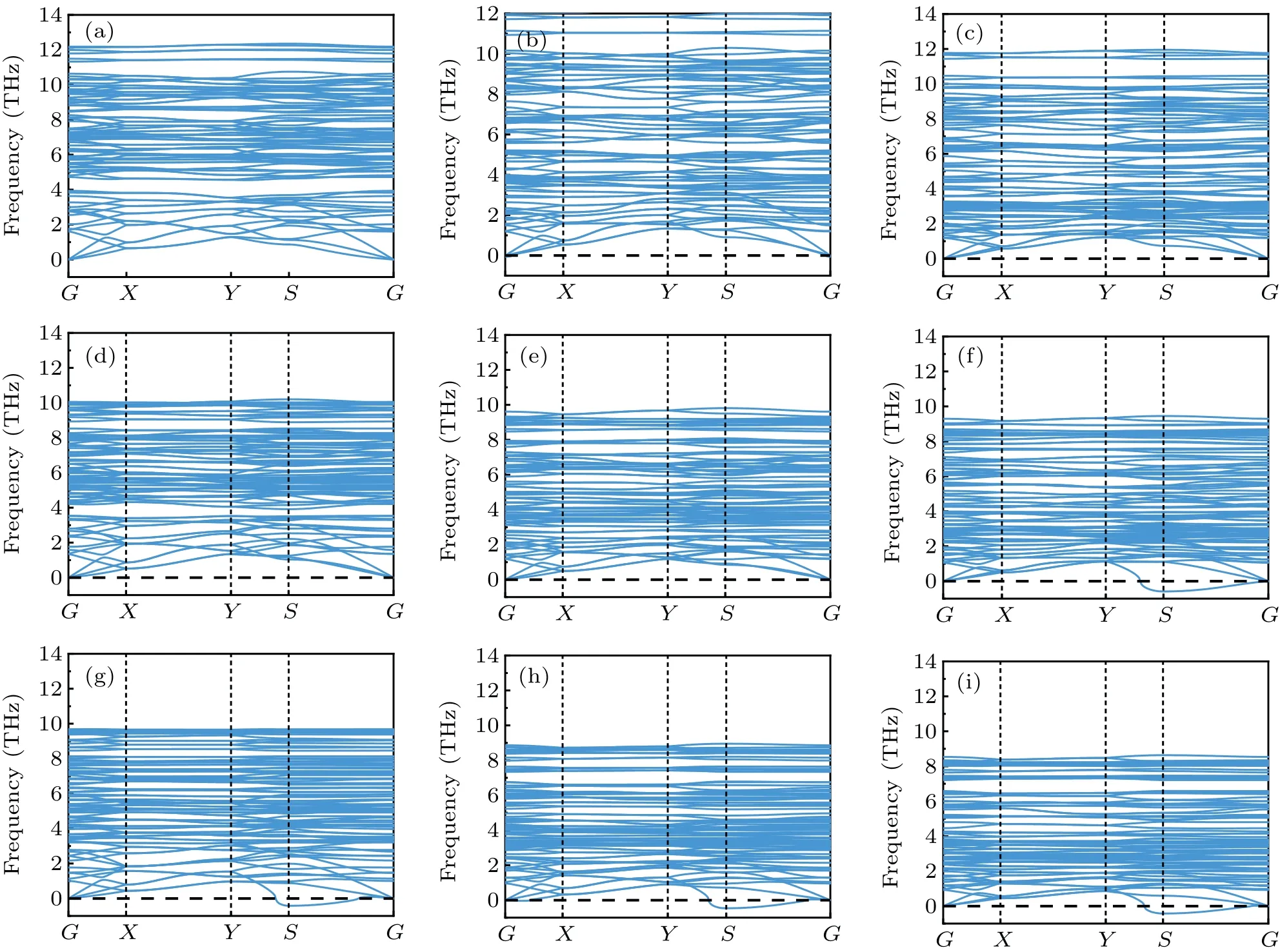
Fig.2.The phonon band structure of monolayer(a)Mo2SCl2, (b)Mo2SBr2, (c)Mo2SI2, (d)Mo2SeCl2, (e)Mo2SeBr2, (f)Mo2SeI2, (g)Mo2TeCl2,(h)Mo2TeBr2,(i)Mo2TeI2.
It is known that the electric and thermal conductivities as well as associated thermoelectric properties severely depend on the chemical bonding characters of the bandedge states.To reveal the chemical bonding characters of the bandedge states,the VBM and CBM states,of Mo2AB2monolayers,Mo2SCl2monolayer is taken as an example to calculate the-pCOHP In Fig.3(b).It can be seen that the CBM state is mainly composed by the antibonding states of Mo-Mo, Mo-S, and Mo-Cl bonds.While the VBM state is formed by bonding state of Mo-Mo bond and antibonding states of Mo-S and Mo-Cl bonds.With increasing atomic numbers ofAorB,weakened antibonding coupling of Mo d-Mo d orbitals tend to reduce the energy positions of the CBM state.As given in Tables 1 and 2,carrier effective masses,meandmhof Mo2AB2monolayer decrease with increasing atomic number ofAdue to the enhanced band dispersion.While from Mo2ACl2, Mo2ACl2,to Mo2AI2,mhandmealong thebdirection increase, which can be ascribed to the decreasing electronegativity from Cl,Br to I.It has been proved that this d-d coupling is responsible for the high carrier mobility and strong anisotropies in mechanical and electronic properties.[32]Of more importance is that the coupled Mo-Mo dimers can enhance the anharmonicity and the scattering of acoustic phonons,which should be beneficial to achieve low lattice thermal conductivity.This unique d-d coupling can be essential to the thermoelectric properties of Mo2AB2monolayer.
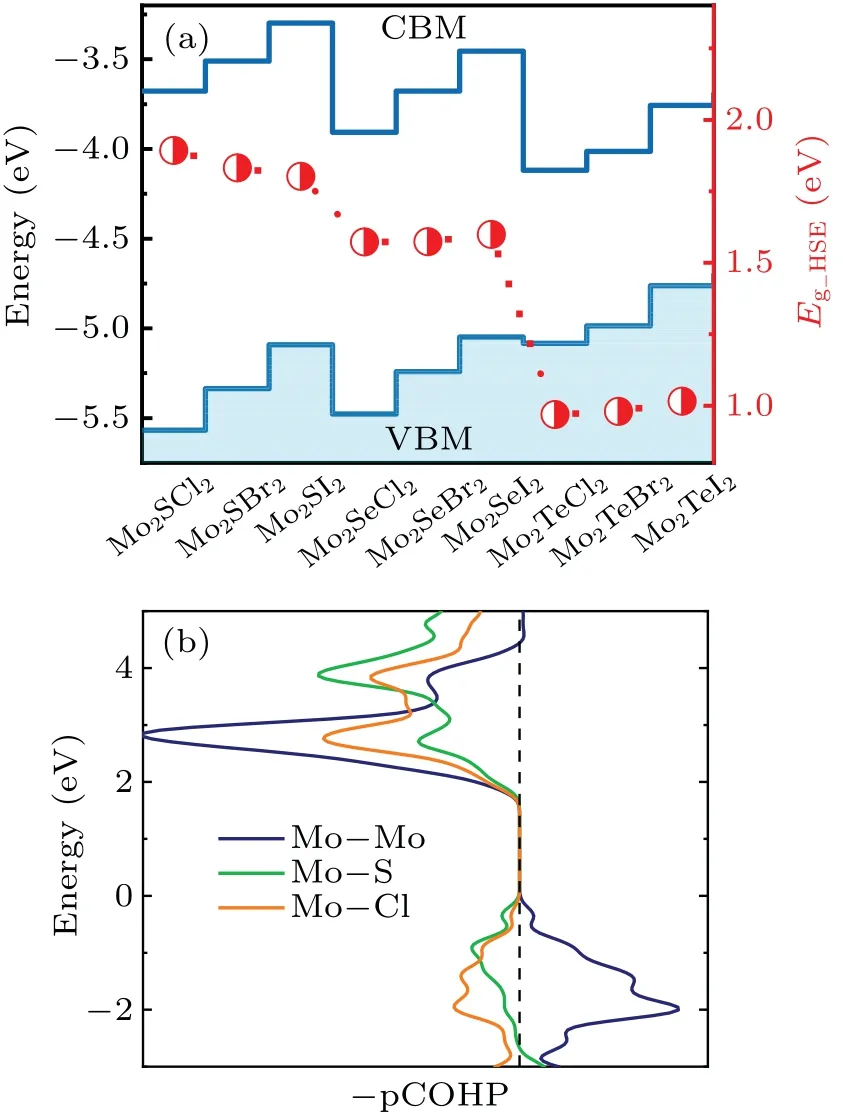
Fig.3.(a) Energy positions of bandedge states and bandgaps of Mo2AB2 monolayers.(b)The-pCOHP analysis for the Mo-Cl,Mo-S,and Mo-Mo bondings in Mo2SCl2 monolayer.-pCOHP values on the left side of the dashed line indicate the anti-bonding states and the those on the right side mean the bonding states.
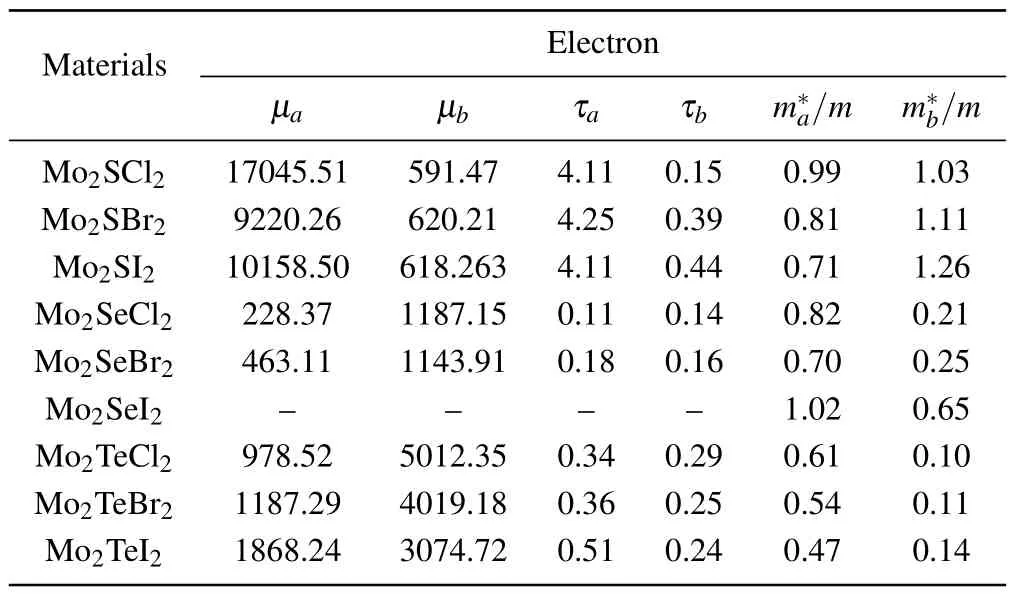
Table 1.The calculated carrier effective mass m∗, carrier mobility µ (in units cm2/(V·s)) and carrier relaxation time τ (in unit ps) along the a and b directions of Mo2AB2 monolayers for electrons. m0 is the static mass of electron.
Insights into electrical transport properties of Mo2AB2can be obtained from carrier mobility (µ), carrier relaxation time(τ),and effective mass(m∗)in Table 1 and Table 2.Our results show that electrons in Mo2SB2and holes in Mo2SeB2and Mo2TeB2have ultra-high carrier mobilities along theadirection and long carrier relaxation time due the weak LA scattering.We confirmed this by calculating the deformation constants in the LA mode,as given in Table S2 in supporting information.Weak LA scattering in Mo2AB2monolayers results in large carrier relaxation time and high carrier mobility,especially along theadirection.According to Eqs.(2) and(3),Mo2AB2monolayers can achieve high electric conductivity and large Seebeck coefficient due to the high carrier relaxation time.Our results indicate that excellent charge transport property and strong anisotropy widely exist in these Mo2AB2semiconductors,which indicates that Mo2AB2can be utilized as optoelectronic and thermoelectricity devices.
As a typical Mo2AB2monolayer,Mo2SCl2monolayer is employed to investigate its thermoelectric performance to provide a comprehensive understanding on the physics of thermoelectric properties of Mo2AB2monolayers.In Fig.4, the direction-dependent electrical transport properties of Mo2SCl2monolayer at different temperatures and carrier concentrations are calculated by solving the BTE, implemented in the BOLTZTRAP code.[45]We find in Figs.4(a)and 4(b)that the Seebeck coefficientSshows no obvious direction-dependent variations and decreases with carrier concentration.At 300 K with carrier concentration of 1012cm-2,|S| of Mo2SCl2monolayer for n-type are 363 µV·K-1in theadirection and 322 µV·K-1in thebdirection, which are much larger than those of SnTe(<150µV·K-1for p-type carriers)[46]and PbTe(<120µV·K-1for p-type carriers).[47]
Figures 4(c) and 4(d), show that the calculated electric conductivities,σ, of Mo2SCl2monolayer increase with the carrier concentration,n.More importantly,for p-type conductivity,σalong thebdirection is much higher than that along theadirection due to the smaller hole effective mass along thebdirection.While in the case of n-type conductivity,electron mobility along theadirection is higher than that along thebdirection.Low scattering rates of the LA mode to electrons give rise to the high electric conductivity, especially in Mo2SCl2along theadirection.
It should be mentioned that electronic thermal conductivity,κe, of these monolayers does not vary by much due to their similar general features in their chemical bonding, following the Wiedemann-Franz law[48](κe=LσT, whereLis the Lorenz factor which can be estimated from the Seebeck coefficient,andL=2.45×10-8W·Ω·K-2.In Figs.4(e)and 4(f),the carrier thermal conductivities,κeof Mo2SCl2are calculated.It is revealed thatκeof Mo2SCl2monolayer as n-type or p-type semiconductor exhibit similar variations as functions ofn.It is found that along theadirection,σandκeof n-type Mo2SCl2are significantly larger than corresponding values of p-type one.These results are in line with the results of carrier mobility and carrier relaxation time in Tables 1 and 2.
In Figs.4(g)and 4(h),the power factor(PF=S2σ)values of Mo2SCl2monolayer as n-type or p-type semiconductor are calculated.Along thebdirection,PFvalues of n-type and p-type Mo2SCl2monolayers exhibit rather similar variations with carrier concentrations with the maximum value of 8.86 mW/mK2.Along theadirection, Mo2SCl2monolayer has much higherPFvalues as an n-type semiconductor than a p-type one due to the larger Seebeck coefficient and electric conductivity along this direction.The maximumPFvalue of n-type Mo2SCl2monolayer at 300 K is 73.85 mW/mK2along theadirection.The highPFvalue implies that Mo2SCl2will have excellent thermoelectric performance.

Fig.4.Calculated thermoelectric parameters of Mo2SCl2 monolayer at different temperatures and carrier concentrations.The p-type (a) and n-type (b)Seebeck coefficient S.The p-type(c)and n-type(d)carrier conductivity σ.The p-type(e)and n-type(f)electronic thermal conductivity κe,the p-type(g)and n-type(h)power factor,PF.
In Fig.5(a), Mo2SCl2monolayer is taken as an example to calculate its detailed phonon dispersion and phonon density of states.It is found that, (i) high-frequency vibrations (>8 THz) are mainly contributed by the covalent Mo-S bondings.(ii) Mo-Cl bondings give rise to vibrations at about 4.5 THz-8.5 THz.(iii) Acoustic phonons that originate from high ionic Cl and Mo ions exhibit weak dispersions.As a result, sound velocity in Mo2SCl2monolayer is relatively low.(iv) The three acoustic modes (TA, ZA, and LA) show direction-dependent dispersions, indicating different sound velocities along theaandbdirections.Anisotropies in lattice thermal conductivity and thermoelectric performance are predicted.(v) Large amount of low-frequency optical vibrations(1.0 THz-4.0 THz)are flat and mainly contributed by Mo atoms with minor contribution of Cl atoms, which correspond to the relative weak d-d bondings of Mo atoms.Additionally, LA and TA phonon modes at the high-symmetrySandXpoints become softened.The thermal conductivity along theaandbdirections will be significantly reduced by these soft modes.Furthermore, these phonon modes show weak dispersions, indicating corresponding vibrations are highly localized and introducing the nearly zero-velocity optical phonon modes.These existing low-frequency localized vibrations have significant contribution to the reduced thermal conductivity.
In addition to the phonon frequency, anharmonicity featured by the Gr¨uneisen parameter,is another important factor in lattice thermal conductivity.In Fig.5(b),Gr¨uneisen parameters of the three acoustic and optical modes are calculated.Our results reveal that ZA modes have large Gr¨uneisen parameters,which means that strong anharmonicity exists,while Gr¨uneisen parameters for LA,TA,and optical modes are much smaller.The strong phonon anharmonicity enhances phonon scattering and greatly reduces thermal conductivity.Further support for the low thermal conductivity of Mo2SCl2monolayer comes from the phonon lifetime calculations, as shown in Fig.5(c).It is revealed that the acoustic modes exhibit much longer lifetimes than optical modes,implying the major roles in the thermal conductivity.Specifically, all the three acoustic phonon have low lifetimes (~10 ps) compared to other results,[29-31]which results in low thermal conductivity.
Then we turn to calculate the lattice thermal conductivity at 300 K,κl, of Mo2SCl2monolayer, as shown in Fig.6(a).It is found thatκldecreases with temperature.At the room temperature (T=300 K), Mo2SCl2monolayer obtains rather lowκlof 1.14 W·m-1·K-1for theadirection and 2.29 W·m-1·K-1for thebdirection,respectively.Figure 6(b)shows the cumulative lattice thermal conductivity along both directions.It is revealed that the acoustic modes make major contribution (75% for theadirection and 69% for thebdirection) toκl.Optical phonon with high frequencies have more influence on the thermal conductivity in thebdirection.The phonon frequencyωiis proportional to the sound velocity in the long-wave limit (or at the center of BZ).Actually, the phonon group velocity indicates the strength of interatomic bonding.In Fig.6(c), the phonon group velocity,Vg, along theaandbdirections are calculated.In the high-frequency range,Vgalong thebdirection is a larger than that along theadirection.While for low-frequency vibration modes,Vgofbdirection are slightly smaller than that of theadirection due to the direct d-d coupling along thebdirection.Therefore,κlin thebdirection is significantly influenced by high-frequency optical modes and is improved by the highVg,despite its small phonon lifetime.Further insights into the anisotropic thermal conduction can be acquired by the calculation of phonon mean free path(MFP),as shown in Fig.6(d).The MFPs for theaaxis andb-axis phonons are 52.04 nm and 132.75 nm,respectively.Longer phonon MFP along thebdirection than that along theadirection indicates reduced phonon scattering rate and enhanced thermal conductivity along this direction,which is consistent with the results in Fig.6(a).These results confirm anisotropic and excellent thermoelectric properties of Mo2AB2monolayers.

Fig.6.(a) The lattice thermal conductivity κl of Mo2SCl2 monolayer as a function of temperature.(b) Cumulative lattice thermal conductivity and(c)phonon group velocity along a and b directions.The grey area in panel(b)represents the frequency range where acoustic modes make major contributions.(d)The variation of cumulative κl versus phonon mean free path for two directions.Temperature is set as T =300 K in panels(b)-(d).
As expected, good anisotropic thermoelectric properties of Mo2SCl2monolayer are confirmed by the calculated thermoelectric figure of merit,ZT, in Figs.7(a) and 7(b).For the case of p-type carriers,ZTin thebdirection is slightly higher than that in theadirection.While for the case of ntype carriers,the situations are just the reverse.In thebdirection,ZTs of n-type and p-type Mo2SCl2show rather similar variations with carrier density.The maximumZTs at 300 K are 0.71 for p-type Mo2SCl2in thebdirection and 3.61 for n-type Mo2SCl2in theadirection.Moreover,ZTs for both n-type and p-type can be significantly improved by increasing the temperature.The n-type Mo2SCl2monolayer exhibits a very highZTlarger than 5 at 700 K along theadirection.

Fig.7.Variations of the thermoelectric figure of merit (ZT) in the a direction(a)and b direction(b)of Mo2SCl2 monolayer as functions of carrier density.
4.Conclusions
In this paper, we employed first-principles approach to explore the electric and thermal transport properties of layered Mo2AB2(A=S, Se, Te;B=Cl, Br, I) materials.Our results reveal that Mo2AB2monolayers can be excellent thermoelectric materials with high electric conductivityσand figures of meritZT.The low lattice thermal conductivities,κl, originate from strong anharmonicities of ZA mode.Direct d-d couplings between Mo atoms,which introduce low-frequency phonon modes, further reduceκland improve thermoelectric performance.Furthermore,the direction-dependent Mo-Abonding and Mo d-Mo d couplings have significant influence on the anisotropies in anharmonicities and transport properties.The thermoelectric properties of Mo2AB2monolayers depend strongly on crystal orientation and carrier density.These results provide rigorous understanding on thermoelectric properties of double-anions compounds and can be very beneficial for designing materials with good thermoelectric performance.
Acknowledgements
Project supported by the Science and Technology Program of Guangzhou City (Grant Nos.202102020389 and 202103030001), the Fund of Guangdong Provincial Key Laboratory of Information Photonics Technology (Grant No.2020B121201011), and the National Natural Science Foundation of China (Grant Nos.11804058 and 12064027).We also thank the Center of Campus Network&Modern Educational Technology, Guangdong University of Technology,Guangdong,China,for providing computational resources and technical support for this work.
猜你喜欢
粮油食品科技(2022年1期)2022-11-24
粮油食品科技(2022年3期)2022-06-01
粮油食品科技(2022年1期)2022-02-11
连环画报(2016年10期)2016-12-16
Water Science and Engineering(2014年1期)2014-03-06

- Chinese Physics B的其它文章
- Single-qubit quantum classifier based on gradient-free optimization algorithm
- Mode dynamics of Bose-Einstein condensates in a single-well potential
- A quantum algorithm for Toeplitz matrix-vector multiplication
- Non-Gaussian approach: Withstanding loss and noise of multi-scattering underwater channel for continuous-variable quantum teleportation
- Trajectory equation of a lump before and after collision with other waves for generalized Hirota-Satsuma-Ito equation
- Detection of healthy and pathological heartbeat dynamics in ECG signals using multivariate recurrence networks with multiple scale factors