
奥拉帕利不同晶型的制备、表征与稳定性研究
2024-01-08 08:13李宏名胡瑞馨郑维江张娇
成都大学学报(自然科学版) 2023年4期
关键词:稳定性
李宏名 胡瑞馨 郑维江 张娇
摘 要:制备奥拉帕利的6种晶型,研究其相对稳定性.采用X-射线粉末衍射分析、热分析、衰减全反射傅里叶红外光谱与拉曼光谱等多种分析检测技术对奥拉帕利 6 种晶型进行了固相表征,并通过高温转晶实验和溶剂介导实验研究晶型间的相对稳定性.奥拉帕利 6 种晶型中1种为水合物晶型H,2种为无水晶型A和L,3种为异质同晶溶剂化物,6种晶型的红外表征数据存在明显差异,可以用于6种晶型的鉴别及生产过程中的晶型控制.X-射线粉末衍射法、差示扫描量热法和拉曼光谱法在晶型A、H与L这3种晶型间存在较为明显的差异,可以用于这3种晶型的鉴别及生产过程中的晶型控制.无溶剂晶型间的稳定性关系为晶型A>晶型 L,晶型 A 为热力学稳定晶型.
关键词:奥拉帕利;多晶型;异质同晶溶剂化物;稳定性
中图分类号:TQ460.6
文献标志码:A
0 引 言
多晶型现象是指一个给定化合物的固态至少存在2种不同的分子排列方式或分子构象,即至少存在2种晶型的现象[1].许多固体药物均存在多晶型现象,原料药不同晶型之间常常存在溶解性、堆密度、吸湿性、压缩性、流动性和稳定性等物理化学性质的差异,这些差异往往会影响原料药的结晶和制剂生产过程,造成制剂溶解性、溶出度和有效期方面的差异,进而影响药物制剂的生物利用度,最终对药物制剂的安全性、有效性和质量可控性造成一定程度的影响.因此,固体药物的多晶型研究已成为药物研发中的重要内容,控制药物晶型是保证药物质量的核心内容和关键环节[2].
奥拉帕利(olaparib,AZD2281) 是阿斯利康公司开发上市的全球首款口服小分子聚腺苷二磷酸核糖聚合酶(PARP) 抑制剂,商品名为利普卓(lynpara),化学名为1-(环丙甲酰基)-4-5-[(3,4-二氢-4-]氧代-1-酞嗪基)甲基-2-氟苯甲酰哌嗪[3].奥拉帕利通过DNA修复基因突变的合成致死作用,靶向作用于癌症细胞,诱导癌症细胞死亡[4],临床主要用于乳腺癌易感基因(BRCA)1/2 突变晚期卵巢癌患者的一线维持治疗.近期的研究结果表明,奥拉帕利对BRCA突变转移性胰腺癌、转移去势抵抗性前列腺癌、三阴性乳腺癌、敏感性小细胞肺癌、不可切除或转移性结直肠癌,以及复发性子宫内膜癌等多种疾病均有临床治疗作用,临床应用前景广阔[5-6].
奥拉帕利属生物药剂学分类系统(BCS)IV类,存在多晶型现象,原研公司上市剂型先为胶囊,晶型A入药,后来为了提高生物利用度将剂型变更为固体分散体,无定型入药,国内暂无仿制药上市.近年来,奥拉帕利的晶型研究在医药领域内受到广泛的关注,目前,文献报道的奥拉帕利晶型主要有无水晶型A、无水晶型L、水合物晶型H、多种溶剂化物和共晶[7-14].本研究采用了多种晶型制备技术,使用二氯甲烷(DCM)、N,N-二甲基甲酰胺(DMF)和乙醇(EtOH)等多种溶剂获得了奥拉帕利的 6 种晶型(晶型A、晶型L、晶型H、DCM溶剂化物、DMF溶剂化物和EtOH溶剂化物),并采用X-射线粉末衍射法(XRPD)、差示扫描量热法(DSC)、热重分析法(TGA)、红外光谱分析法(IR)和拉曼光谱法等多种分析技术对这6种晶型进行了固相表征,并通过高温转晶实验和溶剂介导转晶实验对各晶型的相对稳定性和相互转化关系进行了研究,旨在方便快捷地鉴别奥拉帕利的各种晶型,更加有利于奥拉帕利的质量控制.
1 仪器和试剂
1.1 仪 器
X Pert3 Power型X射线粉末衍射仪(帕纳科公司),DSC2500型差示扫描量热仪(TA仪器公司),TGA1型热重分析仪(梅特勒—托利多集团),Nicolet IS50型光谱仪(赛默飞世尔科技公司).
1.2 试 剂
奥拉帕利粗品(批号为200326,纯度为98.5%),为自制;
无水EtOH(批号为21041402)、DCM(批号为21021901),均购自成都市科隆化学品有限公司;DMF(批号为20201206),购自成都金山化学试剂有限公司.
2 方 法
2.1 多晶型制备
2.1.1 EtOH溶剂化物的制备
取奥拉帕利粗品10 g,加入DCM 90 mL,甲醇10 mL,加热溶解后过滤,于40℃减压蒸除溶剂,得泡状固体(无定型,9.46 g,收率98.9%).泡状固体加入到50 mL无水EtOH中,5℃悬浮 2 h,得白色固体,为EtOH溶剂化物(7.89 g,收率75.9%).
2.1.2 DCM溶剂化物的制备
取奥拉帕利粗品10 g,搅拌下加热溶解于200 mL DCM-EtOH(v/v,1/1)溶液中,趁热过滤,所得滤液于50℃减压蒸除约2/3溶剂后冷却至室温,過滤,固体于40℃干燥,得白色固体,为DCM溶剂化物(9.43 g,收率86.9%).
2.1.3 DMF溶剂化物的制备
取奥拉帕利粗品10 g,搅拌下于90℃加热溶解于30 mL DMF溶液中,趁热过滤,所得滤液重新加热溶解后于60℃析晶,冷却至室温,过滤,固体于40℃干燥,得白色固体,为DMF溶剂化物(9.93 g,收率91.6%).
2.1.4 晶型H的制备
取奥拉帕利粗品10 g,搅拌下加热溶解于50 mL EtOH-水(v/v,1/1)溶液中,趁热过滤,所得滤液缓慢滴加到500 mL纯化水中,滴毕搅拌3 h,过滤,固体于40℃干燥,得白色固体,为水合物晶型H(9.43 g,收率90.9%).
2.1.5 晶型L的制备
取奥拉帕利粗品10 g,80℃加热溶解于120 mL乙腈-水(v/v,3/2)溶液中,趁热过滤,所得滤液于65℃搅拌6 h后冷却至室温,过滤,固体于40℃干燥,得白色固体,为晶型L(8.57 g,收率87.2%).
2.1.6 晶型A制备
取奥拉帕利粗品 15 g,加热溶解于225 mL EtOH-水(v/v,3/1)溶液中,缓慢降温至10℃,过滤,固体在水(130 mL)中加热回流4 h后冷却至室温,过滤,固体于40℃干燥,得白色固体,为晶型A(12.55 g,收率84.9%).
2.2 多晶型表征
2.2.1 XRPD
采用Cu Kα石墨单色器,射线管工作电压为40 kV,工作电流为40 mA,扫描范围为3.5~40.0°,步长为0.013 1°,通过透射式连续扫描方式扫描.
2.2.2 DSC
精密称取上述各晶型样品3~5 mg,均匀铺于盖有穿孔盖的铝坩埚中,加盖扎孔,以空坩埚作空白对照.在氮气保护下升温扫描,温度范围为35~250℃,
升温速率为10℃/min.
2.2.3 TGA
精密称取上述各晶型样品3~5 mg,均匀铺于瓷坩堝中,以空坩埚作空白对照.在氮气保护下升温检测,温度范围为35~250℃,升温速率为10℃/min.
2.2.4 IR
采用金刚石作为衰减全反射(ATR)附件晶体,入射角45°,扫描次数64次,扫 描 范 围为650~3 700 cm-1,分辨率为2 cm-1.
2.2.5 拉曼光谱法
取样品适量,平铺于进样载体凹槽处,置于载物台上,激光功率400 mW,点扫描方式扫描,扫描次数64次,扫描范围为150~3 300 cm-1,分辨率为2 cm-1.
2.3 多晶型相对稳定性研究
2.3.1 高温转晶实验
分别取6种晶型样品各100 mg,于200℃烘箱中加热4 h.
2.3.2 悬浮竞争实验
分别取2种多晶型样品各50 mg,分别加入到1 mL用晶型A饱和的溶剂中,于不同温度下悬浮3 d.
3 结果与分析
3.1 XRPD图谱分析
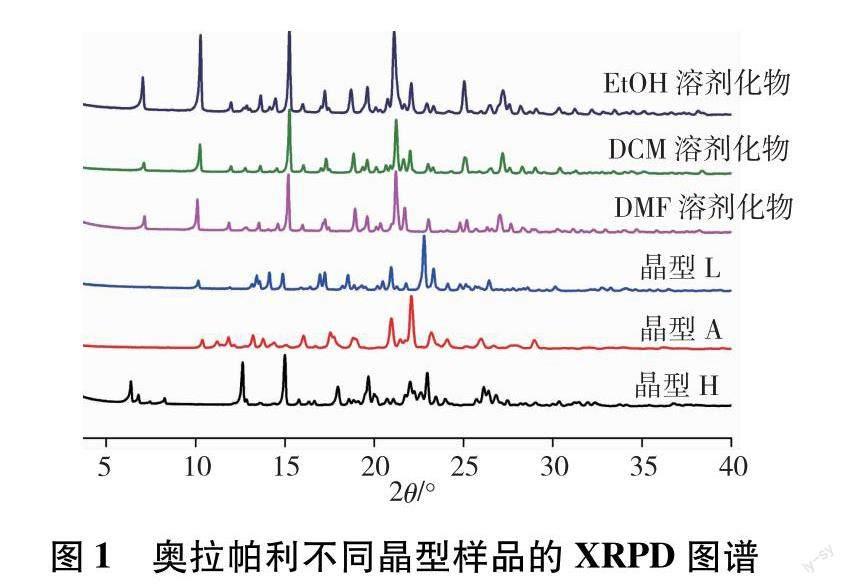
XRPD分析是检测固体产物晶型的最常用手段,常用于药物多晶型的定性和定量分析[15],所得奥拉帕利不同晶型样品的XRPD图谱如图1所示.
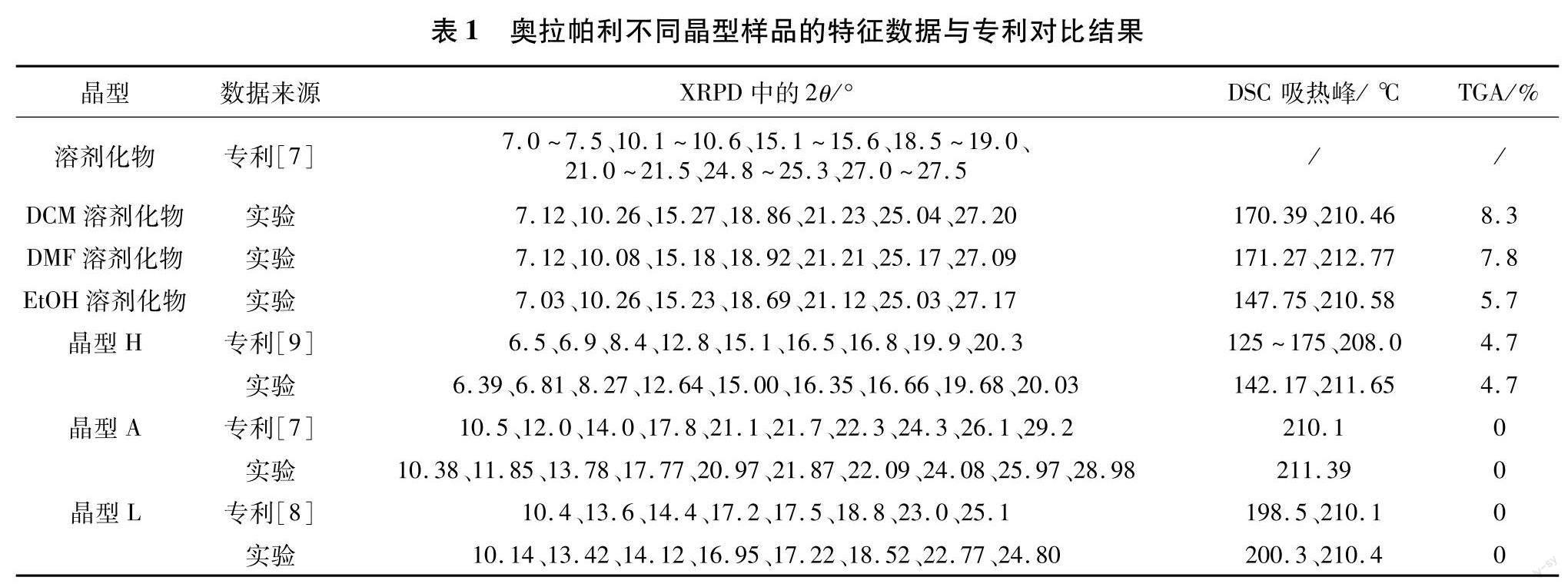
由图1可知,奥拉帕利不同晶型样品的XRPD图谱均有一定的差异.其中,EtOH、DCM和DMF这3种溶剂化物的XRPD图谱比较相似,与文献报道一致,说明这3种溶剂化物结构类似,为异质同晶溶剂化物[16];晶型A、L和H这3种晶型XRPD图谱的衍射峰位置及相对峰强度之间均存在明显的差异,因此可用来判断这3种不同的晶型.晶型A、L和H这3种晶型XRPD图谱的衍射峰实验2θ值与专利数据吻合,不同晶型样品XRPD实验数据与专利数据对比结果见表1.从表中列出的XRPD和热分析数据可以看出,本研究制备得到的6种晶型与专利中报道的晶型一致,且晶型A、L和H的XRPD和DSC数据存在明显差异,可以用于这3种晶型的鉴别.
3.2 热分析
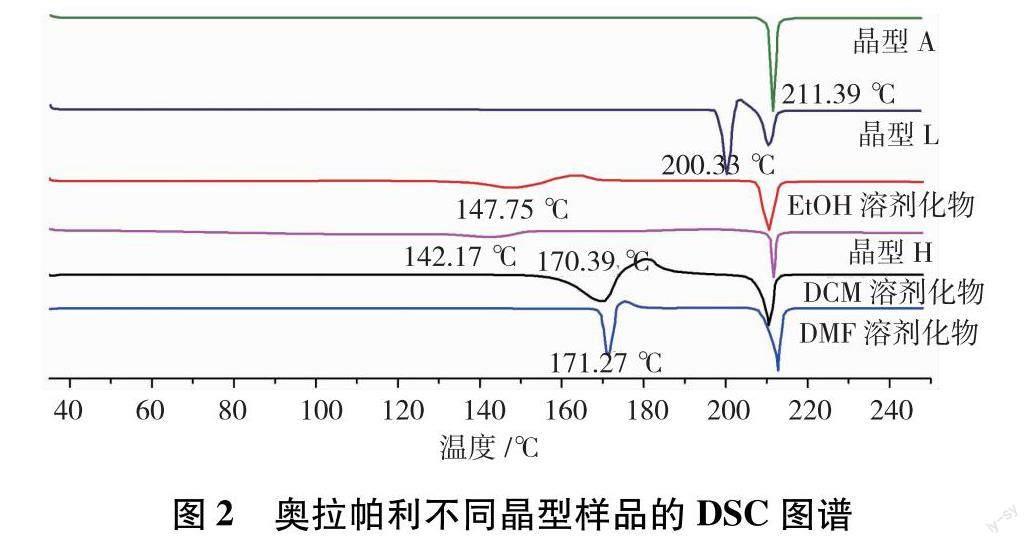
热分析技术常用于药物热分解动力学、药剂成分分析、药物晶型转变与药物晶型鉴别等多个医药领域,已成为药物质量控制的常用技术[17],DSC与TGA是药物研究中最常用的热分析技术.奥拉帕利不同晶型样品的DSC图谱如图2所示.晶型A和L为无水物,晶型A的DSC图谱显示其在分解前的整个升温过程中只有1个吸热熔融峰,峰值为211.39℃;晶型L的DSC图谱显示其在200.33℃吸热熔融,随后放热转晶,于210.49℃再次吸热熔融,说明晶型L在高温下会转化为晶型A,与文献报道一致[8].3种溶
剂化物晶型和水合物晶型H的DSC图谱相似,都是先吸热脱溶剂再放热转晶,最后于210℃左右吸热熔融,说明这3种溶剂化物晶型和水合物晶型H脱溶剂后均转化为无水晶型A.奥拉帕利不同晶型样品的TGA图谱如图3所示.晶型A和L为无水物,250℃前没有失重;晶型H在100℃前失重4.65%,计算可得,晶型H中摩尔比n(水) /n(奥拉帕利)为1,与文献报道一致[9].3种溶剂化物均在120℃以上失重,由失重率计算,可得3种溶剂化物中摩尔比n(溶剂) /n(奥拉帕利) 均为0.5.不同晶型样品DSC与TGA实验数据与专利数据对比结果见表1.
3.3 IR分析
IR是一种方便、快捷的药物分析技术[18].衰减全反射傅里叶红外光谱(ATR-FTIR)作为一种便捷的分析方法,克服了传统FTIR的不足,因其制样简单、能无损测定与检测灵敏度高等优点,已成为分析物质表面结构的一种有利工具和手段,在多个领域中得到了广泛应用,常用于药物晶型的鉴别,奥拉帕利不同晶型样品的ATR-FTIR分析图谱如图4所示.
由图4可知,6种晶型的ATR-FTIR图谱均有差异,ATR-FTIR特征吸收峰见表2.其中,3 700~3 300 cm-1区域,仅水合物晶型H与EtOH溶剂化物有吸收峰,为O-H伸缩振动峰;1 680~1 605 cm-1区域,6种晶型均有3个芳环的特征吸收峰,且峰位置有明显差异,DMF溶剂化物多1个C=O伸缩振动峰;670~650 cm-1区域,仅晶型L有吸收峰,其余5种晶型均没有吸收峰.因此,奥拉帕利不同晶型可用ATR-FTIR方法进行鉴定.
3.4 拉曼光谱分析
IR主要反应分子偶极矩的变化,拉曼光谱主要反应分子极化率的变化,拉曼光谱已逐渐成为鉴别药物不同晶型的重要分析手段.奥拉帕利不同晶型样品的拉曼光谱图谱如图5所示.
由图5可知,6种晶型中,DMF、DCM和EtOH 3种溶剂化物的拉曼光谱图谱差异极小,造成这种现象的主要原因是这3种溶剂化物属于异质同晶溶剂化物,各晶型样品的分子极化率比较类似,因而拉曼光谱图谱也极为相似;6种晶型中,晶型A和L同属于无水物晶型,其拉曼光谱图谱峰形比较相似,仅有几个峰的峰位置差异较为明显;晶型H的拉曼光谱图谱明显与其余5种晶型不同.综合来看,除各种溶剂化物的拉曼光谱图谱比较类似外,其余各种晶型的拉曼光谱图谱均有比较明显的差异,各晶型拉曼光谱图谱的特征吸收峰见表3.3种溶剂化物的特征吸收峰没有明显差异,晶型A、L和H的拉曼光谱图谱的特征吸收峰存在较明显差异,晶型L可选1 451.3、1 486.3和1 501.2作为特征峰,晶型A可选1 444.3、1 481.2和1 494.6作为特征峰,晶型H可选1 441.7和1 479.3作为特征峰,用于鉴别这3种晶型.
3.5 各晶型的相对稳定性
3.5.1 高温转晶实验
分析6種晶型样品于200℃烘箱中高温转晶实验后得到的固体,结果为单一晶型(晶型A),表明晶型A为热力学稳定晶型.
3.5.2 溶剂介导转晶实验
1) 晶型A转为晶型L.晶型A和L在乙腈/水(v/v,1/1)的混合溶剂中60℃下悬浮竞争后,所得固体为单一晶型(晶型L) .
2) 晶型H和3种溶剂化物转为晶型A.晶型A分别和晶型L与3种溶剂化物在乙醇/水(v/v,3/7)的混合溶剂中60℃下悬浮竞争后,所得固体为单一晶型(晶型A).
3) 晶型A和3种溶剂化物转为晶型H.晶型H分别与晶型A和3种溶剂化物在乙醇/水(v/v,3/7)的混合溶剂中20℃下悬浮竞争后,所得固体为单一晶型(晶型H).
4) EtOH溶剂化物转为DCM溶剂化物.EtOH溶剂化物与DCM溶剂化物在 DCM中20℃下悬浮竞争后,所得固体为单一晶型(DCM溶剂化物).
转晶实验表明,奥拉帕利6种晶型在一定条件下可以相互转化,其相互转化关系如图6所示.
4 结 论
本研究对奥拉帕利的6种晶型进行了制备,并通过 XRPD、DSC、TGA、ATR-FTIR和拉曼光谱法对6种晶型进行表征分析.结果表明,6种晶型的ATR-FTIR图谱存在较明显的差异,可以用于6种晶型的鉴别及生产过程中的晶型控制.此外,因获得的3种溶剂化物晶型为异质同晶溶剂化物,其 XRPD和拉曼光谱图谱没有明显差异,只能用ATR-FTIR鉴别,而晶型A、L和H的XRPD、DSC和拉曼光谱图谱存在明显差异,在只有这3种晶型鉴别和控制的情况,也可以采用这3种分析方法.
通过高温转晶实验表明,晶型A是热力学稳定晶型,开发晶型A可以避免生产过程和贮存期间的转晶风险.
参考文献:
[1]高建军,钱帅,高缘.晶型药物研发理论与应用[M].北京:化学工业出版社,2019.
[2]徐悦,吴书平,陈真真,等.依鲁替尼4种晶型的制备、表征与稳定性研究[J].化学工业与工程,2018,35(3):38-42.
[3]范丽萍,焦园园,李然,等.PARP抑制剂olaparib[J].中国新药杂志,2016,25(12):1321-1325.
[4]Slade D.PARP and PARG inhibitors in cancer treatment[J].Gene Dev,2020,34:360-394.
[5]Kindler H L,Hammel P,Reni M,et al.Overall survival results from the POLO trial:A phase III study of active maintenance olaparib versus placebo for germline BRCA-mutated metastatic pancreatic cancer[J].J Clin Oncol,2022,14:JCO2101604-1-JCO2101604-9.
[6]Thiery-Vuillemin A,de Bono J,Hussain M,et al.Pain and health-related quality of life with olaparib versus physician's choice of next-generation hormonal drug in patients with metastatic castration-resistant prostate cancer with homologous recombination repair gene alterations (PROfound):An open-label,randomised,phase 3 trial[J].Lancet Oncol,2022,23(3):393-405.
[7]K·A·米尼尔,A·P·奥特里奇,D·J·隆德斯布拉夫,等.4-[3-(4-环丙烷羰基-哌嗪-1-羰基)-4-氟-苄基]-2H-酞嗪-1-酮的多晶型物:CN201510002348.9[P].2015-05-27.
[8]凯瑟琳·安妮·奎格利,以斯拉·约翰·斯蒂尔,伦纳德·杰西·池亚尔.4-[3-(4-环丙烷羰基-哌嗪-1-羰基)-4-氟-苄基]-2H-酞嗪-1-酮:CN200880111430.3[P].2010-09-01.
[9]M·K·贝赫托尔德,C·B·佩克霍伊泽,J·K·卡希尔,等.药物制剂514:CN200980150172.4[P].2011-11-09.
[10]陈敏华,张炎锋,刘凯,等.奥拉帕尼与尿素的共晶及其制备方法:CN201610235001.3[P].2016-07-13.
[11]陈嘉媚,高璐,戴霞林.一种奥拉帕尼与富马酸共晶晶型α及其制備方法与应用:CN202110925622.5[P].2021-11-12.
[12]Holland J,Eberlin A,Frampton C.Olaparib oxalic acid cocrystals and their pharmaceutical user:WO2022058785(A1)[P].2022-03-24.
[13]李宏名,胡瑞馨,张祥阳,等.PARP抑制剂奥拉帕利的合成及晶型A的制备[J].牡丹江师范学院学报(自然科学版),2022,48(4):45-49.
[14]张娇,黄欣,胡瑞馨,等.奥拉帕利多晶型及晶型转化研究[J].化学工业与工程,2023,40(1):120-126.
[15]Zappi A,Maini L,Galimberti G,et al.Quantifying API polymorphs in formulations using X-ray powder diffraction and multivariate standard addition method combined with net analyte signal analysis[J].Eur J Pharm Sci,2019,130:36-43.
[16]Yang D,Gong N,Zhang L,et al.Isostructurality among 5 solvatomorphs of betulin:X-ray structure and characterization[J].J Pharm Sci,2016,105(6):1867-1873
[17]Monajjemzadeh F,Ghaderi F.Thermal analysis methods in pharmaceutical quality control[J].J Mol Pharm Org Process Res,2015,3(1):1000e121-1-1000e121-2.
[18]刘雯雯,刘晓凤,董航.近红外光谱检测技术快速测定桔梗总皂苷的研究[J].成都大学学报(自然科学版),2021,40(1):20-24.
(责任编辑:伍利华)
Preparation,Characterization and Stability of Different Crystal Forms of Olaparib
LI Hongming1,2,HU Ruixin3,ZHENG Weijiang2,ZHANG Jiao4
(1.College Continuing Education,Wuhan Polytechnic University,Wuhan 430023,China;2.Sinopharm Chuankang Pharmaceutical Co.,Ltd.,Chengdu 611731,China;3.College of Horticulture,Sichuan Agricultural University,Chengdu 611130,China;4.Sichuan Kelun Pharmaceutical Research Institute Co.,Ltd.,Chengdu 611138,China)
Abstract:Six crystal forms of olaparib were prepared and their relative stability was studied.First of all,the forms were characterized by X-ray powder diffraction,thermal analysis,attenuated total refraction infrared spectroscopy and Raman spectroscopy.Then,their relative stability was determined through high-temperature phase transformation and solvent-mediated transformation.Among these Six crystal forms,Form H is a hydrate,Form A and L are anhydrous,and the other three crystals are heteromorphic solvates.Also,there are obvious differences in the infrared spectroscopy data between the six solid forms,which can be used for the identification or polymorph control in the production process.There exist obvious differences in X-ray powder diffraction,thermal analysis or Raman spectorscopy in A,H and L.Furthermore,Form A,L and H can also be identified and controlled by X-ray powder diffraction,thermal analysis or Raman spectroscopy.In addition,the stability relationship is Form A > Form L for solvent-free Forms,that is to say,Form A is the most thermodynamic stable form.
Key words:olaparib;polymorph;isomorphism solvate;stability
猜你喜欢
山东冶金(2022年3期)2022-07-19
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年5期)2021-11-19
石油沥青(2021年4期)2021-10-14
矿产勘查(2020年9期)2020-12-25
数学物理学报(2018年5期)2018-11-16
数学物理学报(2018年1期)2018-03-26
厦门理工学院学报(2016年1期)2016-12-01
现代防御技术(2016年1期)2016-06-01
中国合理用药探索(2014年1期)2014-03-11
