
基于CRISPR/Cas9基因编辑技术构建PD-L1-GFP报告基因HT29细胞株
2024-01-12 12:33谢钰珍覃鸿妮吴凡孙曙光孟丽君
山东理工大学学报(自然科学版) 2024年2期
谢钰珍,覃鸿妮,2,吴凡,孙曙光,孟丽君
(1. 苏州工业园区服务外包职业学院 生物科技学院,江苏 苏州 215123;2.江苏省精准诊疗药物创制工程研究中心(苏州大学),江苏 苏州 215000;3. 苏州东岭生物技术有限公司,江苏 苏州 215123)
PD1(programmed death-1,程序性死亡受体-1)为PDCD1基因编码的Ⅰ型跨膜糖蛋白,主要表达于活化的免疫细胞表面,如T细胞、B细胞、NK细胞以及单核细胞等。PD1是维持自身耐受性的重要因子,在生理条件下可通过TCR识别抗原,调节外周组织中T细胞的功能,从而应对和清除外源病菌及内源异常细胞。PD-L1(程序性死亡配体-1)也称CD274,是PD1的配体之一,是由CD274基因编码的一种细胞表面糖蛋白,多过度表达于肿瘤细胞表面[1]。作为重要的负性免疫调节因子,当T细胞表面PD1受体与肿瘤细胞表面表达的PD-L1配体结合后,可向细胞内传递调控信号,抑制T细胞活化与增殖,从而帮助肿瘤细胞逃脱宿主的免疫监视,因此,抑制PD1/PD-L1通路或者通过特异性靶点抑制PD-L1蛋白的表达,可有效增强肿瘤治疗效果。近年来的临床研究结果显示,由PD1/PD-L1通路介导下的免疫抑制在非小细胞肺癌、胃癌、乳腺癌、大肠癌等多种恶性肿瘤的发生发展中均具有重要作用,PD-L1在以上各种癌组织的表达明显升高,且PD-L1的阳性表达与肿瘤大小、转移情况、分化程度及远期生存率存在密切关系[2-3];此外,从诱导肿瘤细胞表面高表达PD-L1的相关因素来看,目前已报道的肿瘤细胞中调控PD-L1表达的相关信号通路主要有Akt3信号通路和MAPK信号通路、IFN-γ信号通路和AR信号通路[4],而且这些通路的某些抑制剂已被证明可调节PD-L1。例如:TGFβR-1抑制剂LY364947可明显降低TGFβR-1诱导的PD-L1mRNA和蛋白表达[5];突变转录因子FOXP3在PD-L1启动子区的结合位点后,胰腺癌细胞PD-L1的表达明显受到抑制[6];但关于结肠癌肿瘤细胞PD-L1表达的具体调控机制仍需进一步研究。CRISPR/ Cas9系统是一种新型基因编辑技术,通过sgRNA介导核酸酶Cas9对基因组特定位点进行识别、切割,从而实现基因编辑,操作相对简便,基因编辑效率高。本文利用CRISPR/Cas9技术,在PD-L1基因终止密码子之前插入绿色荧光蛋白GFP基因,通过构建稳定表达PD-L1-GFP报告基因的结肠癌细胞株HT29,旨在建立一个可直观观察细胞上PD-L1是否表达以及表达强弱的系统,为进一步研究结肠癌细胞中PD-L1表达的可能调控机制提供帮助,为后期体内外筛选调控PD-L1的上游新靶点及药物奠定基础。
1 材料与方法
1.1 材料
HT29细胞和293T细胞(苏州东岭生物技术有限公司保存),Lenti CRISPR V2质粒载体和pUC19载体(苏州东岭生物技术有限公司),胶回收试剂盒(Thermo),Stbl3感受态大肠杆菌(TransGen Biotech),DMEM培养基和D10培养基(HYclone),T4连接酶(Takara公司),DNA聚合酶(NEB)。
1.2 方法
1.2.1 sgRNA引物的设计与合成
根据NCBI查询的PD-L1基因序列,使用CRISP 在线设计工具(http://www.e-crisp.org/E-CRISP/),根据靶点设计原则,在PD-L1基因终止密码子处设计sgRNA,并在序列正义链与反义链的5’端添加Esp3I(BsmBI)酶切位点,合成的具体序列如下:
sgRNA-1: GAGGAGACGTAATCCAGCAT
gRNA-1-F:CACCGAGGAGACGTAATCCAGCA
gRNA-1-R:AAACATGCTGGATTACGTCTCCTC
sgRNA-2: GTCTCCTCCAAATGTGTATC
gRNA-2-F:CACCGTCTCCTCCAAATGTGTATC
gRNA-2-R:AAACGATACACATTTGGAGGAGA
为检测突变是否成功,设置了一对检测引物,扩增产物为包含sgRNA靶点的基因组DNA片段,检测引物具体序列如下:
Test-F: TGGGGGACAAGCCATCCCAA
Test-R: ATGATTTGCTTGGAGGCTCC
1.2.2 LentiV2-gRNA重组质粒的构建及鉴定
根据所设计序列合成单链sgRNA,经梯度降温PCR退火形成双链sgRNA。退火结束后,将得到的双链gRNA连接到已用Esp3I酶切回收后的线性Lenti-V2空载质粒中,连接产物转化Stbl3感受态细胞,37 ℃培养过夜后挑取阳性单克隆进行测序验证,测序引物:LKO1-5(GACTATCATATGCTTACCGT)。针对序列正确的单克隆,提取其对应菌液中的质粒DNA,即可得到正确的Lenti-V2-sgRNA重组质粒。
1.2.3 pUC19-Donor-GFP重组质粒的构建及鉴定
设计左同源臂+GFP+右同源臂序列,序列两端添加酶切位点XbaI/BamHI,送公司合成Donor片段,将合成的Donor片段、pUC19分别用XhaI和BamHI双酶切后连接。取10 μL连接产物转化至50 μL Stbl3感受态大肠杆菌中,37 ℃培养1 h后,均匀涂在含有Amp的LB平板上,过夜培养。次日挑取6个克隆于4 mL含有Amp的LB培养基中,37 ℃培养16 h。16 h后取1 mL菌液抽提质粒,并对质粒进行菌落PCR,判断选取的单克隆中是否含有目的片段。将验证正确的质粒送至测序,测序引物:M13-R (CAGGAAACAGCTATGACC),测序正确后冻存菌种。
1.2.4 细胞培养和细胞转染
从-80 ℃冰箱中取出冻存的HT29细胞,复苏结束后留2 mol/L的细胞量在10 cm培养皿中培养,隔天进行传代培养。培养至第4天时将HT29转移至24孔板中的2孔(一孔转染,一孔阴性对照),每孔0.5×106细胞。转染前1~2 h更换新鲜培养基(0.9 mL/孔),按Lenti-V2-sgRNA(0.6 μg)、Donor-GFP(0.4 μg)、1 mg/mL PEI(3 μL)、DMEM(97 μL)配制转染体系,室温孵育30 min后,将混合液加入到1孔中,另一个孔无须加入,轻轻摇匀后放入培养箱培养(37 ℃,5%CO2)。
1.2.5 多克隆效果验证
提取转染后的HT29细胞基因组DNA,利用在PD-L1基因组以及GFP上分别设计的上下游引物,对其进行PCR鉴定,以检测其中是否含有在PD-L1位点成功插入GFP片段的目的细胞。引物序列:PD-L1-F(TTCAAATTTATCATTTATCA),GFP-R(CCGGACACGCTGAACTTGTG),PCR体系:模板DNA10ng、PD-L1-F和GFP-R各1μL,2X PrimeSTAR HS DNA Polymerase MIX 10 μL,总体积20 μL。PCR扩增程序:98 ℃预变性20 s,98 ℃ 变性 10 s,55 ℃退火5 s,72 ℃延伸35 s,共30 个循环,68 ℃ 彻底延伸 2 min。扩增的PCR产物经1%琼脂糖凝胶电泳分析。
1.2.6 单克隆细胞筛选
转染72 h后,观察细胞生长状况并计数,采用有限稀释法,用D10(10%的FBS+DMEM)按每200 μL一个细胞稀释到96孔板中,培养24 h后,显微镜观察荧光及细胞状态,并做好标记。继续培养,当上一步标记的单克隆细胞密度达到80%后,消化并收集细胞,用基因组DNA抽提试剂盒提取基因组,作为检测引物(TestF、R)的扩增模板,最后将扩增产物送至公司测序,以检测PD-L1-GFP报告基因是否插入成功。
2 结果与分析
2.1 Lenti-V2-gRNA重组质粒构建结果
重组质粒基因测序结果显示,在酶切位点之间插入的片段位置、方向以及序列与预期一致(图1),含有本实验所需要的两条sgRNA序列,证明LentiV2-gRNA重组质粒构建成功。
2.2 两条sgRNA切割效果验证结果
为了验证所设计的两条sgRNA的切割效果,将两种Lenti-V2-gRNA重组质粒转入293T细胞后提取基因组DNA,并对其进行T7E1酶切鉴定,结果显示1和2两种sgRNA都能切下相应大小的条带(图2),证明Lenti-V2-sgR1和Lenti-V2-sgR2都具有切割效果,本实验随机使用其中1条,采用 sgRNA2,即Lenti-V2-sgR2。

图1 Lentiv2-gRNA重组质粒测序结果

图2 T7E1酶切图
2.3 pUC19-Donor-GFP重组质粒构建结果
以挑选的6个单克隆为模板,经菌落PCR均能扩增出目的片段(图3),说明菌落中含有目标质粒。将阳性质粒进一步送至金唯智测序,从图4可以看出,第一行的序列为测序结果,第二行的序列是所需的模板序列,序列比对完全正确,pUC19-Donor-GFP质粒构建成功。

注:1-6为随机挑取的6个单克隆,M为DNA marker。图3 pUC19-Donor-GFP重组质粒转化质粒菌落PCR结果

图4 pUC19-Donor重组质粒测序结果
2.4 Lenti-V2-sgR2和pUC19-donor-GFP细胞转染结果
将Lenti-V2-sgR2和pUC19-donor-GFP转染至HT29细胞,72 h后荧光显微镜下观察细胞状态。由图5可知,培养72 h后可观察到少量细胞表达绿色荧光,证明GFP成功转入HT29并进行了表达。
2.5 PD-L1-GFP报告基因工程细胞株阳性单克隆筛选结果
2.5.1 多克隆效果验证结果
图6为多克隆PCR鉴定结果,可以发现实验组在200 bp位置有目的条带,而对照组没有,证明GFP插入到指定位点。
2.5.2 单克隆细胞筛选结果
为将上述验证的插入正确GFP序列位点的细胞挑选出来,将多克隆细胞进行单克隆筛选,图7为荧光显微镜观察结果,可以看到细胞形成了单克隆并且具有均一的荧光,可以进行后续的验证。

(a)细胞图

(b)荧光图图5 HT29细胞荧光蛋白表达

图6 多克隆验证琼脂糖凝胶电泳图

图7 单克隆细胞荧光状态图
为验证上述挑选的单克隆中所需的序列存在,对其基因组PCR之后送至金唯智测序,图8显示与正常HT29细胞基因组相比,敲入的实验组所挑的单克隆在终止密码子前插入了GFP片段,证明细胞系构建成功。
3 讨论
PD1表达于活化的免疫细胞表面,如B淋巴细胞、CD4+T细胞等。PD-L1作为PD1的配体之一,在癌组织上可见异常高表达,如肾癌、肝癌、肺癌、乳腺癌及卵巢癌等;但在肿瘤邻近的正常组织中低水平表达,提示它参与肿瘤发生发展,并在削弱抗肿瘤免疫反应中发挥重要作用。
PD1/PD-L1是负性共刺激分子,当肿瘤细胞表面的PD-L1与免疫细胞表面的PD1结合时,可抑制免疫细胞的增殖与活性甚至诱导其凋亡,使癌细胞成功逃脱免疫杀伤。近年来,PD-L1及其受体PD1信号通路一直是肿瘤免疫领域的热门研究对象,针对阻断PD1/PD-L1通路的单克隆抗体已然成为了肿瘤免疫治疗的明星产品,目前FDA 已经批准了五种PD-L1抑制剂。作为非特异性免疫治疗产品,PD-L1抑制剂的抗癌效应具有广谱性,且在不同癌症疾病中显示出了很好的治疗[7-11],但由于治疗过程中的原发性和获得性耐药,相当大比例的患者无法从中受益。相较于单药疗法,近年来越来越多的研究正向着PD1/PD -L1耐药机制研究以及联合用药靶点的筛选上转移[12]。
在一些研究报道中,针对不同类型免疫检查点的联合疗法已被证明对几种肿瘤有效。例如:采用抗PD-1抑制剂抗体、抗CD137激动剂抗体和疫苗治疗的三联疗法可以显著增强胰腺导管腺癌的治疗效果[13-14];联合anti-PD-L1和anti-TIGIT在临床上对转移性NSCLC患者非常有效[15];增强 ITCH 活性可促进 PD-L1泛素化降解从而降低肿瘤细胞 PD-L1 表达,化合物AK087 与 MAPK抑制剂联用可明显增强 MAPK 靶向治疗黑色素瘤的效果[16]等。但对于其他大部分肿瘤来说,不同疗法治疗过程中肿瘤表面PD-L1的表达量变化情况及产生治疗抗性的关键机制,仍需进一步研究。
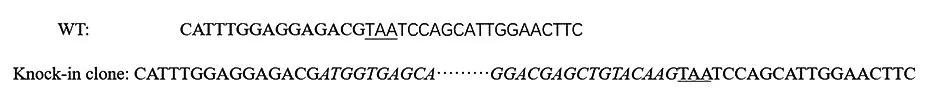
注:斜体为GFP片段,下划线为PDL1终止密码子,WT为正常HT29测序序列,Knock-in clone为实验组测序序列。图8 对照组和实验组序列对比图
为了探究结肠癌细胞表面PD-L1表达的更多可能调控机制,本研究利用CRISPR/ Cas9系统和同源重组的原理,通过两次转染将GFP荧光基团成功插入到 PD-L1基因终止密码子之前,成功构建了PD-L1-GFP报告基因HT29细胞株,通过荧光信号直观观察细胞上PD-L1是否表达以及表达的强弱程度,为进一步研究结肠癌细胞中PD-L1表达的可能调控机制提供了材料,并为后期体内外筛选调控PD-L1的上游新靶点及药物奠定了基础。
猜你喜欢
今日农业(2021年11期)2021-08-13
中国现代医药杂志(2020年10期)2020-12-14
食品科学(2018年10期)2018-05-23
现代检验医学杂志(2016年3期)2016-11-15
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
中国当代医药(2015年9期)2015-03-01
西南军医(2015年6期)2015-01-23
遗传(2014年3期)2014-02-28
