
亨廷顿舞蹈病的临床特点与基因诊断(附一个家系报告)
2012-05-25 07:49何瑜玢,夏莉,梁静静
中国神经精神疾病杂志 2012年7期
亨廷顿舞蹈病又称亨廷顿病(Huntington's disease,HD)、慢性进行性舞蹈病,是一种影响运动功能为主的神经退行性疾病,以基底节及大脑皮层神经元缺失为基本病理特征。IT15(interesting transcript 15)基因是HD的致病基因,定位于4p16.3,是由于该处外显子1的CAG重复序列的异常扩展突变引起[1]。我们收集了湖北一个临床拟诊为亨廷顿舞蹈病的家系,采用DNA直接测序方法检测IT15,以提供遗传咨询以及探讨IT15基因中CAG的重复拷贝数与发病年龄、临床特征之间的关系并综述亨廷顿舞蹈病治疗的最新进展。
1 资料与方法
1.1 对象
一经临床拟诊为 HD家系(图 1),对该家系进行详细的临床病史调查并尽可能全面收集患者的临床资料包括实验室检查、脑电图、头颅CT、MRI和简易智力状态量表(mini-mental state examination,MMSE),都经过临床检查排除了其他内外科疾病。按知情同意和自愿参与原则,该家系中有11名成员参与(包含2名有症状成员)。
1.2 研究方法
1.2.1 提取 DNA 采集肘静脉血 4 mL,应用DNA提取试剂盒提取外周血白细胞基因组DNA,DNA提取试剂盒购于上海赛百盛基因技术有限公司。
1.2.2 PCR扩增 ①上游引物序列(t15f):CCCAGAGCCCCATTGATTGCC;下游引物序列(t15r):GGCGGCGGTGGCGGCTGTTGC,引物由上海生工生物工程技术服务有限公司合成;②以受试者外周血白细胞基因组DNA为模板进行PCR扩增,反应体系为10X的PCR缓冲液(含Mg2+)5μL,2.5mM的dNTPs4μL,10μM引物t15f和t15r各2μL,基因组DNA(100ng/μL)4μL,5U/μL的热启动TaqDNA聚合酶(TAKARA)0.5μL,加水补足50μL;③应用9700型自动扩增仪(美国ABI公司)进行扩增反应,反应条件为预变性94℃,8min,(94℃,30s;64℃,30s;72℃,30s)为1个循环,共40个循环,最后72℃,延伸5min。

图1 HD家系图谱,即先证者
1.2.3 电泳 将PCR扩增产物各取5μL进行2%琼脂糖电泳检测。
1.2.4 基因测序 将PCR产物直接测序,测序引物采用t15f,并对有明显双特异性条带的扩增产物,进行切胶纯化后测序。
2 结果
2.1 临床特点 该家系共28人(家系图谱见图一),6例患者,男3例,女3例,首发年龄在35~71岁,平均54.3岁;已逝的3例患者病程9~15年,平均11.3年。呈父系遗传,第Ⅰ代起病年龄71岁,第Ⅱ代平均起病年龄59岁,第Ⅲ代平均起病年龄39岁:先证者46岁,43岁时出现轻微左肩耸动,目前已进展至吐词欠清,双肩耸动,左侧肢体不自主运动,以上肢为主。神经系统查体主要为四肢肌张力低,四肢腱反射活跃,双上肢Rossolimo′s和Hoffman′s 征(+),双下肢踝阵挛(+),指鼻和跟膝胫试验完成不协调。家属诉其记忆力下降,暴躁易怒伴抑郁倾向,表现为语言减少,不爱活动。Ⅱ9:62岁起病,病程已10年,起初表现为一侧腿抬高,逐渐进展为全身不自主运动伴认知障碍,目前查体:不能行走,神清,计算力下降,反应迟钝,严重构音障碍,吞咽困难,不自主吐舌头,嘴角牵动,全身不自主运动,双手徐动,全身肌肉重度萎缩,四肢肌力V级,四肢肌张力高,四肢腱反射减弱,指鼻和跟膝胫试验不能,家属诉患者性格固执,舞蹈样动作减少;据家属介绍Ⅰ1和Ⅱ3与Ⅱ9临床表现类似,Ⅱ3病历中记载病程中服用氟哌啶醇和安坦片期间曾有好转,最终因呼吸循环衰竭死亡;Ⅱ1起初表现为双肩耸动,双上肢扣扣子样不自主动作,病情发展也与Ⅱ9类似;Ⅲ5:35岁起病,以双下肢抖动为主,不倒地。
2.2 主要实验室特征 先证者脑电图检查未见明显异常;Ⅱ3病历中记载头颅CT、MRI示脑萎缩;Ⅱ9MMSE评分小于10分。
2.3 电泳结果 2例已发病患者(Ⅱ9和Ⅲ4)和1例无症状成员(Ⅳ1)均有特征性两条扩增带,一条含正常(CAG)n重复数,约220bp,另一条含异常扩展的(CAG)n重复,约300bp左右;余8名成员只显示一条扩增带,约220bp(图2)。
2.4 基因测序 对所有成员的PCR产物直接测序,其中对有明显双特异性条带的扩增产物,进行切胶纯化后测序;3名成员CAG重复数目大于等于40:Ⅱ9为17/40,Ⅲ4为17/45,Ⅳ1为16/48,其他8名成员CAG重复数目在20次以下。具体见图3和表1。

图2 PCR后电泳结果注:Marker:50 bp梯度相对分子质量标准;Ⅲ4和Ⅱ9为有症状患者,Ⅳ1为症状前患者,余为健康成员
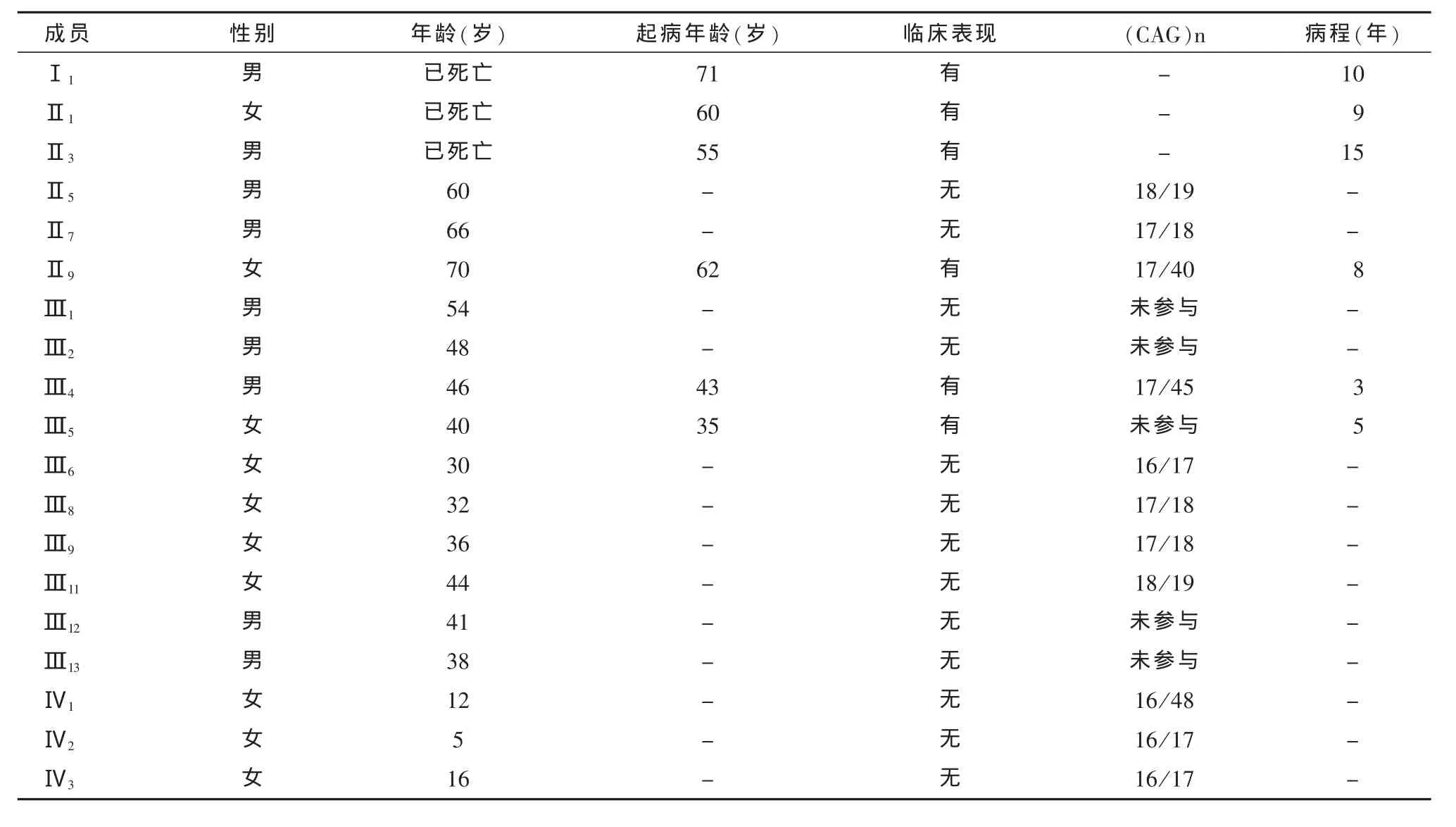
表1(CAG)n与起病年龄和临床表现之间的关系
3 讨论
亨廷顿舞蹈病多中年起病,以慢性进行性舞蹈样动作、认知障碍和精神行为异常三联征为典型特点,呈迟发型常染色体显性遗传的神经变性疾病,一般病程在15~20年左右[2],临床上头颅CT和MRI提示基底节区脑萎缩,无特异性,确诊须靠IT15基因检测。国外通过对欧美大样本人群IT15基因检测分析得出[3]:CAG重复数≤26时未发现引起疾病;在27~35时未发现引起疾病,但可引起突变,在减数分裂时不稳定,易发生扩展突变,尤其是父系遗传时;在36~39时为不完全外显的HD等位基因,携带者可能发病也可能不发病,≥40时为完全外显的HD等位基因,携带个体必然会患病。我们收集的这个家系起病年龄平均54.3岁,舞蹈样动作逐渐加大,其中4例晚期伴智能障碍、性格固执和脾气暴躁,基因测序结果显示有症状的2例患者CAG重复数目大于40;IT15基因检测结果为杂合子,结合家系图谱中代代遗传,中年起病的特点表明该家系呈迟发型常染色体显性遗传,故可明确诊断该家系疾病为亨廷顿舞蹈病。其中Ⅳ1为女性12岁,目前无任何临床表现,但电泳显示特征性两条扩增带,基因测序CAG重复数目大于40,我们称之为症状前患者,该患者必然发病,但起病时间目前不能确定,并有50%的可能将IT15基因遗传到下一代,故将来孕早期建议行产前诊断。
通过家系图谱,我们可以看出该家系呈父系遗传,且每代发病年龄较上代提前,体现出遗传早现的特点,这与国外报道一致[4];有文献报道,(CAG)n重复越多,发病越早,累计50%~70%的符合这个规律[5],其他不符合的情况可能与基因变异和环境有关;但(CAG)n重复长度与病情进展的速度无显著的关联[6],在我们收集的这个家系里,Ⅱ9CAG重复40次,62岁发病;Ⅲ4,45次,43岁发病,一定程度也提示了发病年龄与CAG重复数呈负相关。Ⅳ1,48次,目前未发病,其发病年龄有待观察;这3例结果也提示随着年龄的增加,逐渐表现出症状,可能与IT15基因表达的亨廷顿蛋白聚集有关。有学者提出HD的发病年龄可能与MSX1基因[7]、ApoE基因[8]、ADORA2A[9]基因或Atg7基因上的V471A的多态性[10]等有关。
目前关于HD的治疗,致力于减慢疾病进展,药物均处于试验性阶段,仍未发现特异性药物[11]。目前辅酶Q10[12]、利鲁唑[13]、艾斯能[14]以及肌酸[15]这四种药物有研究报道在治疗HD有一定疗效,但仍需长时间大样本的随机对照试验证实,其中辅酶Q10和肌酸已进入临床试验Ⅲ期。有学者认为HD非常适合作为神经保护药物的模型,可能推迟甚至阻止其临床表现的发生[16]。提倡HD从无症状阶段开始干预可能对预后有利,HD治疗前景还是很乐观的[2]。
致谢:感谢潘松青教授对我的课题指导和论文修改,感谢这个家系成员的合作,感谢夏莉,梁静静帮助我收集资料,感谢北京尤比爱生物科技中心杨福辉对我们收集的家系成员DNA进行基因测序。
[1]Huntington′s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington′s disease chromosomes[J].Cell,1993,72(6):971-983.
[2]Walker FO.Huntington′s disease[J].Lancet,2007,369(9557):218-228.
[3]The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group.ACMG/ASHG statement.Laboratory guidelines for Huntington disease genetic testing[J].Am J Hum Genet,1998,62(5):1243-1247.
[4]Kutuev IA,Khusainova RI,Khidiyatova M,et al.Analysis of the IT15 gene in Huntington′s disease families[J].Russian J Genetics,2004,40(8):919-925.
[5]Wexler NS.Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington′s disease age of onset[J].Proc Natl Acad Sci USA,2004,101(10):3498-3503.
[6]Rosenblatt A,Liang KY,Zhou H,et al.The association of CAG repeat length with clinical progression in Huntington disease[J].Neurology,2006,66(7):1016-1020.
[7]Djoussé L,Knowlton B,Hayden MR,et al.Evidence for a modifier of onset age in Huntington disease linked to the HD gene in 4p16[J].Neurogenetics,2004,5(2):109-114.
[8]王育新,张本恕,张伟.ApoE基因对亨廷顿病发病年龄的影响[J].中风与神经疾病杂志,2010,27(7):615-617.
[9]Elahe TF,Carsten S,Stefan W,et al.Age at onset in Huntington’s disease:replication study on the associations of ADORA2A,HAP1 and OGG1 [J].Neurogenetics,2010,11(4):435-439.
[10]Silke M,Meiju S,Hong VC,et al.Age at onset in Huntington′s disease is modified by the autophagy pathway:implication of the V471A polymorph-hism in Atg7 [J].Hum Genet,2010,128(4):453-459.
[11]Gardian G,Browne SE,Choi DK,et al.Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington′s disease[J].J Biol Chem,2005,280(1):556-563.
[12]Huntington Study Group.A randomized,placebo-controlled trial of coenzyme Q10 and remacemide in Huntington′s disease[J].Neurology,2001,57(3):397-404.
[13]Landwehrmeyer GB,Dubois B,de Yebenes JG,et al.Riluzole in Huntington′s Disease:A 3-Year,Randomized Controlled Study[J].Ann Neurol,2007,62(3):262-272.
[14]de Tommaso M,Difruscolo O,Sciruicchio V,et al.Two years′follow-up of rivastigmine treatment in Huntington disease[J].Clin Neuropharmacol,2007,30(1):43-46.
[15]Beal MF.Neuroprotective effects of creatine[J].Amino Acids,2011,40(5):1305-1313.
[16]Ross CA,Tabrizi SJ.Huntington′s disease:from molecular pathogenesis to clinical treatment[J].Lancet Neurol,2011,10(1):83-98.
猜你喜欢
广西林业科学(2022年6期)2023-01-16
区域治理(2022年40期)2022-11-27
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
科学24小时(2019年4期)2019-06-10
医药前沿(2019年29期)2019-01-05
科学生活(2016年7期)2016-07-25
