
3种化学键离解能的区别和联系
2012-09-25 03:39罗渝然
大学化学 2012年2期
罗渝然
(中国科学技术大学化学与材料科学学院 安徽合肥 230026)
1 大一学生提出的问题
我们使用《普通化学原理》(第3版)[1]做大一学生的参考教材。在答疑课时,一位学生问:该书第278页的下部注释如何理解?为什么氢分子中的H—H键(离解)能有3个数值(458,432和436kJ/mol)?我的回答是:这一现象有普遍性,任何分子都有此特性,你们的知识还不够,先承认它吧。进入高年级课程,答案自然会有。
我的回答是“想当然”。因我长期旅居海外,不熟悉国内现行各大学教材之间的衔接。但随后,我查阅了物理化学、量子化学、结构化学等许多版本的国内教材。令我惊讶的是,这些教材并没有提供上一问题的答案。我认为,它是各专家在为本科生“编织多维知识网”(编写各分支学科大学教材)时的一处遗漏。
学生要理解上一问题,需要有分子振动、零点能、分子转动、化合物等压热容、热力学标准条件和非标准条件下化合物焓变等预备知识。这些知识分别属于分子物理学、物理化学、化学热力学、统计热力学、结构化学、量子化学、光谱学和质谱学等分支学科。国内某些物理化学、量子化学、光谱学及材料学的教材,讨论过De和Do之间的区别与联系。但没有任何大学教材讨论De,Do,D2983者之间的区别与联系。这确实遗憾。建议专家在编写新教材时,关注分支学科之间相互交叉、相互渗透、又很有实用价值的教学内容。
2 我们的论述
为了理解De,Do,D2983者之间的区别与联系,首先要搞清De,Do,D298的不同定义。文献[2]和文献[3]论述了这一问题。De,Do,D298这3个物理量有相同点,也有不同点。相同点是三者都讨论1mol理想气体的RX发生化学键离解反应(1)的能量变化。

(1)
这里,R与X是碎片(原子、分子、自由基或离子等)物种,RX是其缔(化)合物。
De,Do和D298是离解反应(1)对应不同温度下物理量,各有不同的明确定义,如图1所示。
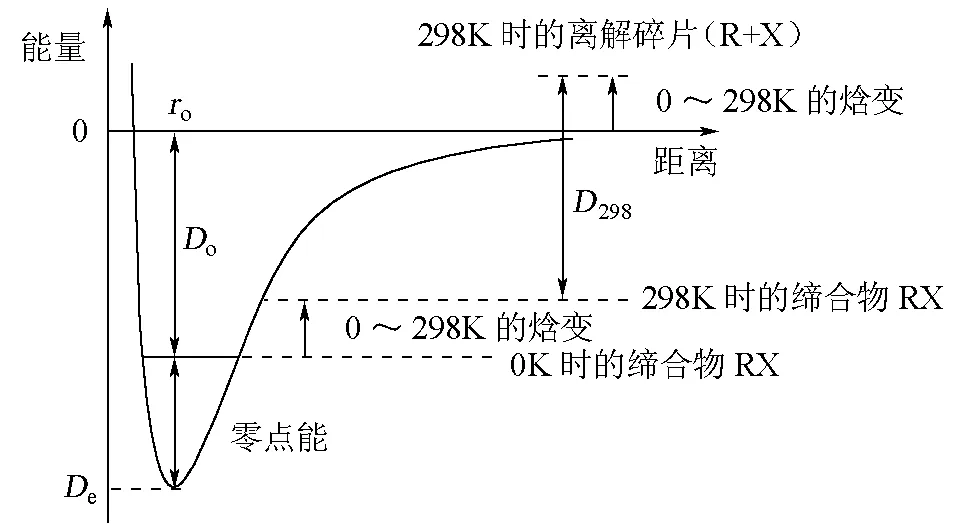
图1 De,Do和D298的区别和联系[2-3]
图1中De是0K时的纯理论的键(离解)能,源于量子化学计算,很难用实验技术直接测量。此物理量又叫碎片物种(R+X)之间在0K时的结合能(binding energy)。
通常,碎片物种(R+X)与缔(化)合物RX在不断地平动、转动和振动。但在0K时,平动和转动自由度冻结。碎片物种(R+X)与缔(化)合物RX的振动能之差,叫零点能(zero point energy,ZPE)。如图1所示,Do和De通过零点能相关:
Do=De-Δ(零点能)
(2)
这里,Δ(零点能)=零点能(R)+零点能(X)-零点能(RX)。等式右端3项,分别对应于多原子碎片与缔合物中的零点能。对于双原子分子(或离子),零点能的计算特别简单:

(3)
这里,h是Planck常数,ν和ωe分别代表双原子分子(或离子)中的基态振动频率和光谱常数。
化学键的键(离解)能D298定义为在热力学标准条件下,1mol理想气体RX中化学键R—X发生离解反应(1)时的焓变。对化学键均裂过程,按Hess定律,得:
D298(R—X)=ΔfHӨ(R) + ΔfHӨ(X)-ΔfHӨ(RX)
(4)
这里,ΔfHӨ(R),ΔfHӨ(X)和ΔfHӨ(RX) 分别代表碎片和缔合物是理想气体时的标准生成热(焓)。
与Do明显不同,D298(R—X)是298.15K时的物理量。温度从0K上升到298.15K,多原子碎片和缔合物中许多自由度被激发,这分别相应于碎片和缔合物从0K到298.15 K的焓变。从图1看到,D298(R—X)就是298.15K时,产物(R+X)与反应物RX的标准生成热的差值,即式(4)。
表1的前4栏总结了De,Do和D298的区别,包括相应温度、对研究化学过程的重要性以及数据来源等。对于研究实际的化学反应,D298的概念远比De和Do更有价值也更加重要,测量方法也多得多(见表1第4栏),已积累的D298实验数据近两万个(见表1第5栏)。

表1 De,Do和D298的区别
顺便提一句,本短文不涉及平均键能。因为曾经有过历史功绩的平均键能法在今天其实用价值已经丧失。关于这一问题,笔者已在另处讨论[5]。
3 双原子分子的键离解
对于双原子分子(或离子),De、Do和D298的区别和联系很简单。按分子物理学中的能量均分定律,等压条件下,离解反应(1)前后的等压热容改变为:
ΔCp=R/2+R=3R/2
(5)
从0K到298.15 K,等压热容改变对焓变的贡献是ΔCp×(298.15-0)。因此,Do和D298的关系是:
(6)
下面以氢分子的H—H键离解为例,详细叙述De(H—H),Do(H—H)和D298(H—H)的区别和联系。
首先,计算氢分子中的零点能:

这里,ωe的数值取自文献[4]。
2004年,张彦鹏和他的同事[6]用高分辨脉冲激光光谱技术测量获得:
Do(H—H)=36118.062±0.010cm-1(432.0619±0.0001kJ/mol)
这一测量值的精度非常高,远远超过“化学精度”(±4.184kJ/mol),进入了“光谱精度”(±1cm-1或±0.0120kJ/mol)。根据这测量值,我们可推算:
De(H—H)=Do(H—H)+零点能=432.0619+26.3251=458.3870±0.0001kJ/mol
以及氢分子的H—H键能测量值[2-3,7]:
D298(H—H)=Do(H—H)+3.71805=435.7799±0.0001kJ/mol
表1的右边最后栏,归纳了根据张彦鹏等[6]观测的Do(H—H)推算出来的De(H—H)和D298(H—H)数值。
4 为什么手册上很难查阅到De和Do的数值?
在大型工具书“CRCHandbookofChemistryandPhysics”中[7],笔者列举了约3700个典型双原子和多原子物种(分子、离子、自由基、络合物等)中的化学键的D298数值,Dr.Lide介绍了大约270个双原子分子的De值,该大型手册没有直接给出Do值。在文献[3]中,笔者提供了近2万个化学键的D298数值。这本最新的化学键能数据大全,也不提供De和Do的数值。为什么手册上很难查阅到化学键的De和Do的数值? 主要理由有以下两点:
1) 对研究实际的化学反应,D298数值远比De和Do的数值更有价值。De和Do是绝对0度时的物理量。0K下,分子的平动自由度被冻结,分子之间没有碰撞,分子不可能发生化学反应。这叫化学第零定律[8-9]。换句话说,只有在温度大于0K,化学反应才发生。De和Do等绝对0度时的物理量,有理论上的重要性,而对实际化学反应仅有参考价值。反之,D298数值是热力学标准状态下的物理量。D298与其他热力学标准状态下的其他物理量互相紧密关联,例如自由原子和自由基的标准生成热(焓)。虽然几乎所有的化学反应不是在热力学标准状态下发生,但是按化学热化学的规律,可预测实际条件下(远偏离热力学标准状态)的化学过程。这些转换细节,在化工热力学的教材中有丰富的文字说明。
2) 观测化学键的D298值的方法要比测量Do的方法多很多,积累的化学键的D298测量数据已近两万[3,7,10],而Do的测量数据相对很少。正因为这原因,化学出版物逐渐形成一条不成文的习惯约定,如果在文章中没有特殊强调和文字交代,提到某化学键的键离解能的数值,就是指D298值。但是在光谱学和质谱学的文献中,某化学键的键离解能的数值,通常是指Do值,即直接从实验获得的值。
在收集、选择、评论、构建化学键能数据库时[2-3,7,10],考虑到上面提及的习惯约定和绝大多数读者的要求,我们已经把从光谱和质谱测量获得的Do数据转换成D298值,即没有同时列出Do和D298值。如果某些读者希望知道当初观测的Do数据,可查阅数据库中[3,7,10]提供的原始文献。
总之,目前大学教材仅论述De和Do之间的区别与联系,忽略了在讨论化学反应时更有实际价值的D298。有人对国内教材给出如下评论:当前教材多考虑自身理论系统的完整性 (这颇像自耕农在几亩土地内多年辛勤地精耕细作),不仅与现代科学中的实际问题联系很不够,而且经常避开相互交叉、相互渗透、灵活与多角度的科学课题。本文为该评论观点提供了又一个实例。
5 精确观测的键能数值将冲击精确的量子化学计算
众所周知,目前任何量子化学计算(分子轨道法,密度泛函法)均非严格的,都需要输入若干可靠实验数据(其中包括自由原子的标准生成热(焓))用于选择或调整。因为精确的键能观测值可推算出自由原子的标准生成热(焓),故它有可能冲击精确的量子化学计算。下面看一个实际例子。2005年,莫宇翔小组利用离子速度成像法[11],给出:
Do(F—F)=1.606±0.001eV(154.952±0.096kJ/mol)
Do(F+—F)=3.334±0.001eV(321.674±0.096kJ/mol)
F—F和F+—F都是双原子物种,利用式(6),我们推算出热力学标准条件下的键能:
D298(F—F)=158.670±0.096kJ/mol
D298(F+—F)=325.393±0.096kJ/mol
根据Hess定律和键(离解)能的定义,我们推算出氟原子的标准生成热(焓):
ΔfHӨ(F)=D298(F—F)/2=79.335±0.068kJ/mol
这个由莫宇翔小组2005年测量结果导出的氟原子的标准生成热比CODATA(Committee on Data for Science and Technology,国际科学技术数据委员会)在1984年推荐的、至今仍被文献接受的79.38±0.30kJ/mol更精确。我们建议,在用量子化学方法研究氟化合物的化学反应时,最好采用更精确的氟原子的标准生成热(焓),ΔfHӨ(F)=79.335±0.068kJ/mol。基于后一数据的量子化学计算,将使含氟化合物的理论预测更精细化。
6 结论与建议
(1) 国内化学教材在为本科生编织多维知识网时,遗漏了一个重要的内容,即没有讨论3种化学键离解能(De,Do,D298) 的区别和联系。
(2) 本短文论述了3种化学键离解能(De,Do,D298) 的区别和联系,并以氢分子的H—H键离解为例,给出了详细的解释。
(3) 在实际化学反应中,描述化学键断裂和生成的D298的概念和数据远比De和Do更有实用价值,因此,建议把3种化学键离解能(De,Do,D298) 的区别和联系,增补到新版的物理化学或有关大学教材之中。这有利于灵活、多角度教学,使大学生了解键能的准确概念(化学中的最基本概念之一),以及它的广泛应用。
参 考 文 献
[1] 华彤文,陈景祖,严洪杰,等.普通化学原理.第3版.北京:北京大学出版社,2005
[2] 罗渝然,郭庆祥,俞书勤,等.现代科学中的化学键能及广泛应用.合肥:中国科学技术大学出版社,2008
[3] Luo Y R.Comprehensive Handbook of Chemical Bond Energies.USA:CRC Press,2007
[4] Haynes W M.CRC Handbook of Chemistry and Physics.92nd ed.USA:CRC Press,2011
[5] 罗渝然,俞书勤,张祖德.大学化学,2011,26(4):67
[6] Zhang Y P,Cheng C H,Kim J Y,etal.PhysRevLett,2004,92:203003
[7] Luo Y R.Bond Dissociation Energies∥CRC Handbook of Chemistry and Physics.92nd ed.USA:CRC Press,2011
[8] 罗渝然,俞书勤,张祖德,等.大学化学,2010,25(4):78
[9] 罗渝然,Benson S W.化学通报,1989(10):22
[10] 罗渝然.化学键能数据手册.北京:科学出版社,2005
[11] Yang J,Hao Y,Li J,etal.JChemPhys,2005,122:134308
猜你喜欢
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
电子制作(2016年19期)2016-08-24
考试周刊(2016年60期)2016-08-23
制冷技术(2016年4期)2016-08-21
考试周刊(2016年48期)2016-06-29
中学生数理化·高二版(2016年6期)2016-05-14
莫愁·家教与成才(2016年4期)2016-04-13
科普童话·百科探秘(2015年7期)2015-07-25
发明与创新·中学生(2015年8期)2015-07-21
汽车与新动力(2014年4期)2014-02-27
