
亨廷顿舞蹈病:教学性综述
2015-11-02 06:34JeanMarcBurgunder
中国神经精神疾病杂志 2015年10期
Jean-Marc Burgunder
·专家论坛·
亨廷顿舞蹈病:教学性综述
Jean-Marc Burgunder*

Jean-Marc Burgunder,欧洲亨廷顿病协作网主席(European Huntington’s Disease Network,EHDN),瑞士亨廷顿中心负责人(Swiss Huntington’s Disease Center),瑞士伯尔尼大学神经科教授(University of Bern,Switzerland),中国亨廷顿病协作网执行委员会委员,四川大学华西医院、中南大学湘雅医院及中山大学附属第一医院客座教授。
亨廷顿舞蹈病(Huntington’s disease,HD)是一种罕见的常染色体显性遗传病。患者一般在中年发病,出现运动,认知和精神方面的症状。亨廷顿舞蹈病的病因是由于亨廷顿基因上多核苷酸重复序列的错误表达,从而影响不同的分子通路,最终导致神经功能失调和退化。现在此病可由基因检测确诊,不过在检测前,必须进行慎重的神经遗传咨询。由于亨廷顿舞蹈病目前只能对症治疗,治疗须综合考虑患者及其家人的要求。此综述将简要概述亨廷顿舞蹈病的临床病症,病理生理及治疗方法。
1 引言
亨廷顿舞蹈病(Huntington’s disease,HD)是一种全外显性的常染色体显性遗传病。它的临床症状复杂多变,常累及运动,认知和精神三个方面。患者病情呈进行性恶化,通常在发病15~20年后死亡。近年来,亨廷顿研究组(Huntington Study Group,HSG)以及欧洲亨廷顿病协作网(European Huntington’s Disease Network EHDN)等研究机构的不懈努力及大量临床研究,大大提高我们对此病的认识。与此同时,亨廷顿病基因的发现,使我们能够利用细胞与动物模型对其机制进行研究,从而使我们对其发病机制的有深入了解。此综述将以教学为目的,主要阐述亨廷顿舞蹈病临床表现,病理生理学机制及其治疗手段方面的基本知识。
2 临床表型
亨廷顿舞蹈病(HD)的主要临床症状通常分为三大类,包括运动症状,认知功能障碍及精神障碍。除此外,HD也会累及到其他系统,其他系统症状可为原发性也可为继发性的并发症[1]。HD发病一般在40岁左右。不过,也有非常年轻或非常年长发病的案例,这些患者通常出现不典型的临床症状,比如儿童患者多表现为严重发育迟缓[2],而老年患者的症状可能非常轻微[3]。
HD症状的评估分两阶段:第一阶段应在首次检查时进行,包括全身及神经系统的体检,重点检查HD相关的舞蹈,认知和精神体征,力求得出全面准确的疾病诊断及分析。第二阶段是在发病过程中进行,检查应着重于确认及了解患者及其家人的主诉,以便提供治疗缓解症状。在某些观察性研究中,两阶段的症状评估可以根据已设定的指南标准相应进行。这些研究包括由欧洲亨廷顿病协作网组织的EHDN Registry Study以及Enroll-HD项目。
HD的典型症状是舞蹈症(表1)。长期以来,这一疾病相应地被命名为亨廷顿氏舞蹈症(Huntington’s Chorea)。然而,舞蹈症并不是HD患者身上出现的唯一运动障碍,在一些患者中非舞蹈症状会比舞蹈症状更加严重影响功能。因此,目前标准命名是亨廷顿病(Huntington’s disease,国内大部分文献及教材目前仍使用原译名,既亨廷顿舞蹈病)。
舞蹈症本身类似于左旋多巴引起的运动障碍。运用多传感磁定位系统作全身记录的研究发现舞蹈症与左旋多巴引起的运动障碍,除了运动速度存在细微差别外,其他参数非常相似[4]。评估舞蹈症症状很重要的一点是系统性评估从而排除其他潜在病因,这一点对于舞蹈症症状不典型和没有HD家庭病史的患者特别重要(表2)。
HD的认知症状(表1)有时会比舞蹈症早很多年出现。有研究发现,HD基因携带者中的40%有轻度认知功能障碍,且越接近发病其认知障碍越明显[5]。这些早期变化严重影响患者功能,此类早期功能退化可以由患者的运动,认知和抑郁症状预测[6]。HD患者认知功能障碍的类型与其他神经退化疾病,如帕金森氏或阿尔茨海默氏病的类型不同。在HD疾病的早期,认知功能损害主要局限于单个脑功能域而不累及整个脑部[5]。在疾病早期,患者不仅加工速度和情节记忆会受到影响,执行功能障碍也比较明显[7]。患者也可能有情感认知,特别是厌恶认知方面的缺陷。这可能严重妨碍他们的社会交往能力。执行功能障碍也是HD的一个主要问题[8]。HD患者往往无法认识到症状的严重程度,一些患者甚至出现出病感失认。随着病情的发展,患者痴呆症状常常恶化,不过即使是病情严重的案例,也仍可能保存部分认知功能。
HD的精神症状(表1)常常复杂多变。反映出复杂并相互作用的不同神经心理机制,这些机制直接与疾病过程中的脑功能损伤相关。早在运动症状出现前,HD患者就有可能由于知悉家庭病史和疾病风险而产生焦虑,并可出现抑郁发作。随后,患者也可能会出现性格方面的轻微变化,并出现攻击性行为和去抑制行为。而自杀也是HD的一种常见并发症。
受疾病影响,HD患者会有睡眠障碍[9],如睡眠潜伏期延长,睡眠效率下降,夜间觉醒次数增加及深慢波睡眠减少。以上描述的部分症状是HD特有,与其他神经退行性疾病无关[10]。睡眠状况的改变与其他临床症状以及脑形态学变化的严重程度密切相关。但是,在发病早期,患者似乎并未察觉这方面的变化[11]。
患者在HD发病后,通常要历经几个阶段。从家庭病史开始,患者早在青春期就对HD有了解。患者对自身是否是携带基因产生困扰,并开始对出现的症状和不同程度的功能障碍产生恐惧。HD症状在发病后进行性恶化,通常在发病15~20年后最终导致患者死亡[12]。
医师不仅要做有关诊断的临床检查,而且必需在疾病过程中对病情进行精确的评估和记录。采用这样一种对症论治的方法,就能及时了解病患的需求,从而根据患者的需要提供具体的治疗方法。HD综合评估量表(Unified Huntington’s Disease Rating Scale,UHDRS)中包括了对运动,功能,行为和认知症状的评定[13],其评定方法有准确的指引,包括原版影象和网络资源(www.euro-hd. net,为EHDN注册会员提供)[13]。
3 疾病诊断
详细了解患者病史并进行体检后,通常就能知道运动障碍的成因并能对运动障碍分类[14]。因此全面收集患者不同方面包括家族史的信息十分重要(表3)。特别需要准确了解家族几代人的病史,特别是有血缘关系的亲人。某些隐患还需注意,包括病患和亲属间的非亲子关系。病患还可能由于社会、心理或其他因素而对病情有所隐瞒。体检的目的是确定运动障碍的症状和特点,这也有助于发现其他方面的病症,包括感觉、锥体和神经肌肉的受累情况。

表1 亨廷顿舞蹈病临床表型

表2 鉴别诊断舞蹈症状时的其他潜在病因
HD为常染色体显性遗传病,隐匿起病,症状包括舞蹈动作,认知和行为障碍进行性加重,症状表现在同一家族的成员身上可能会有所不同。一旦确定有家族史,HD的可能性就非常大,确诊HD则不需更多的检测。不过如果患者家族中没有任何成员曾经做过基因检测,还是可以考虑为患者做基因检测确诊[15]。由于HD不单只波及患者更会影响到其家人,进行基因检测前一定要慎重进行遗传咨询,咨询最好是能让患者家人参与,同时必须充份考虑到患者的接受程度。虽然在人群中,特别是西方人群中,患者具有典型的临床表现而基因检测却为阴性的可能性很小[16],但如果出现这种情况,则需要更多的检查指导进一步的诊断(表2)。许多表现出舞蹈症症状的散发性疾病都可以通过准确的病史和身体检查确诊。通常相对简单的实验室检查就有助于进一步的确诊(表3)。某些特殊的患者可能还需要进行影像学和神经生理学检查(表3)。
4 流行病学调查
HD的患病率在不同人群中变异很大,西方人口的患病率是十万人中有4~8例[17]。最近一个荟萃分析显示中国/日本HD患病率为0.40,而欧洲/北美以及大洋洲患病率为5.7[18]。在中国文献中以前只有少数的个案报道[19]。然而,随着中国亨廷顿病协作网的发展,协作网中的各中心已接触到越来越多的病例,相信HD患病率在未来会有更证。
5 疾病进程
大多数情况下,HD患者已从自己家人方面对HD有了深刻了解。在年幼时,他们可能就已经认识到在自己家族中有这样一种疾病。渐渐,患者亲属,特别是患病者的孩子,会对自身患病的风险产生疑问。之后,他们为了确定,可能会考虑基因检测。如果患者被证实携带这一变异基因,他们随后很长一段时间可能都无法确定自己的一些轻微改变是否就已经发病。患者慢慢可能在工作和生活上开始出现困难,他们的亲人也会发现患者在情绪和行为上发生了细微变化。在之后的一些年里,患者的身体机能逐年减弱,到最后将需要重症监护,这将为其家人带来沉重的负担。随着疾病程度不断加深,身体机能如进食,括约肌的控制也都会逐渐退化。同时,由于构音障碍,沟通也越来越困难。最终患者往往死于肺炎。而在疾病早期,自杀也并不罕见。
6 遗传学
HD是一个常染色体显性遗传病。在确认基因位点后,又经过多年研究搜寻才找到它的致病基因Htt(Huntingtin)[20]。这个基因编码Htt蛋白(Huntingtin protein),在它的第一个外显子中,包含了重复的CAG三联密码子。在HD中,这个三联密码子的重复次数会出现异常增加。拥有多于36次CAG重复三联子的个体就会患病。但如果重复次数为36~39个,则全外显性较低。三联子重复次数不稳定,在遗传到下一代时次数可发生改变。
基因检测特别是对仍未发病的潜在基因携带者的检测必须慎重[21]。必须根据咨询者自身情况,提供高质易懂的咨询信息,让患者能自己作出决定。基因检测的一个主要风险是可能出现自杀倾向。特别对于有精神病史及基因检测前已经失业5年以上的患者,自杀风险会增加[22-23]。
7 病理机制
HD患者的主要病变部位是在尾状核和豆状核,然后是大脑皮层(主要是在前叶),以及内侧苍白球、丘脑、下丘脑[24-25]。这些病变会导致严重的全脑萎缩,与同龄正常对照组相比较,脑体积减少高达30%。早在确诊发病的多年以前,就可以检测到尾状核的变化和脑体积的减少[26]。

表3 HD诊断程序
在大脑中表达的变异Htt蛋白通过不同分子机制导致神经功能退化。变异蛋白不仅能促使该蛋白的异常功能增加而且能导致正常功能丧失。虽然多年来我们就发现了变异Htt蛋白会产生积聚[27],然而对有关蛋白积聚是否对神经元有害或者蛋白积聚仅是对变异Htt蛋白毒性产生的保护作用目前尚未有定论[25]。正常Htt蛋白本身具有多种细胞功能,Htt蛋白变异则导致这些功能障碍。蛋白变异常常首先表现为相关基因的表达异常,已有研究显示HD纹状体中有关神经传导的基因出现表达异常[28-29]。另外,CAG的异常重复可以大规模影响分子间的相互作用,导致细胞内蛋白运输紊乱[30]。例如与线粒体形态和功能稳定相关的蛋白相互作用的紊乱,常导致能量供应不足和神经退化[31]。Htt蛋白变异不仅能打乱线粒体功能相关蛋白的基因调节,而且还可与和线粒体膜表面的蛋白反应,损伤呼吸链功能,妨碍线粒体固定到微管,影响线粒体动态融合与分裂并使钙传输增加。变异蛋白也可抑制自噬功能,促进凋亡,改变神经营养供能及细胞胞浆内的生物和信号合成。
8 治疗方法
现有的几种HD疗法可以归类为对症治疗或对因治疗。对症治疗的目的是根据医生客观诊断和患者主观回馈信息,直接缓解患者病症。对症治疗还可以进一步分类。根据针对运动、精神和认知方面的不同的主要症状的治疗,可分为HD特异和HD非特异对症治疗。
对因治疗则包括直接的基因疗法和其他间接的分子疗法。前者把变异基因和它的转录产物作为唯一的病因,直接进行治疗;后者的目的是更正导致疾病的复杂分子和神经相关通路。虽然对因治疗目前还无法实现,但针对上述不同的分子通路,正在开展大量的研究,以期减缓疾病[32]。
除此之外,我们还必须重视HD患者的护理以支持其家人。由于针对患者的需要进行症状治疗非常重要,最好能通过多学科合作来实施。在发病前和疾病第一阶段,这个多学的团队可以包括神经专科医生、精神科医生和遗传咨询师,随着患者病情的变化,则需要理疗师、语言和职业治疗师、护理人员及其他专业医疗人员的加入。而临终关怀和末期处理也越来越受到重视[33]。由于只有少量的HD临床治疗试验供参考[34],有关HD对症治疗的的建议的证据相当缺乏[35]。因此,我们必须依靠专家意见,有时也要或多或少地根据针对其他疾病的舞蹈和精神症状的研究来探索HD的治疗方式。治疗的主要目的仍然是改善患者的生活质量。但即使是针对临床表现最明显的运动障碍,有关的治疗数据也十分有限[36-37],因此,我们最近已开始了治疗舞蹈症状药物在世界各大州应用的调查[38]。根据患者的需要,如果出现包括耻辱感、身体损伤,步态不稳,工作困难和睡眠不安等症状,则表示需开始服用治疗舞蹈症的药物。服用这类药物有不同的药物选择(表4)。当患者合併出现精神病或攻击行为时则首选精神类药物。丁苯喹嗪是美国食品药品局(FDA)批准治疗HD舞蹈动作的药物之一[39]。丁苯喹嗪的主要机制是抑制单胺囊泡转运(vesicular monoamine transporter,VMAT)。其用药量达到100 mg/d时,可以显著减轻舞蹈症状[40],但如果停止用药,舞蹈症状会加重[41]。使用此类药物须注意其副作用,包括运动迟缓伴震颤。如果是使用丁苯喹嗪,则还需注意抑郁症。精神类药物还可能包括奥氮平(2.5~10 mg),利哌利酮(0.5~2 mg),或泰必利(50~200 mg)。注意用药时需要根据患者反应调整剂量。对于一些特定的患者,有时可能需尝试几种不同的精神类药物,从而找出最合适的。如果出现肌阵挛,可使用2-丙基戊酸钠[42]。巴氯芬和苯二氮卓可对疾病末期的运动障碍治疗有效。而化学神经阻滞剂如肉毒杆菌毒素注入高度兴奋的肌肉中则可治疗局灶痉挛(focal spasms),如磨牙症(bruxism)或局灶性痉挛反射亢进(focally predomi-nant spastic overactivity)。同时,物理治疗也很重要。近期研究显示,重点训练姿势和步态对HD患者有很大帮助[43]。最近,巳出版了为患者疾病不同阶段提供物理治疗的初步指南[44]。
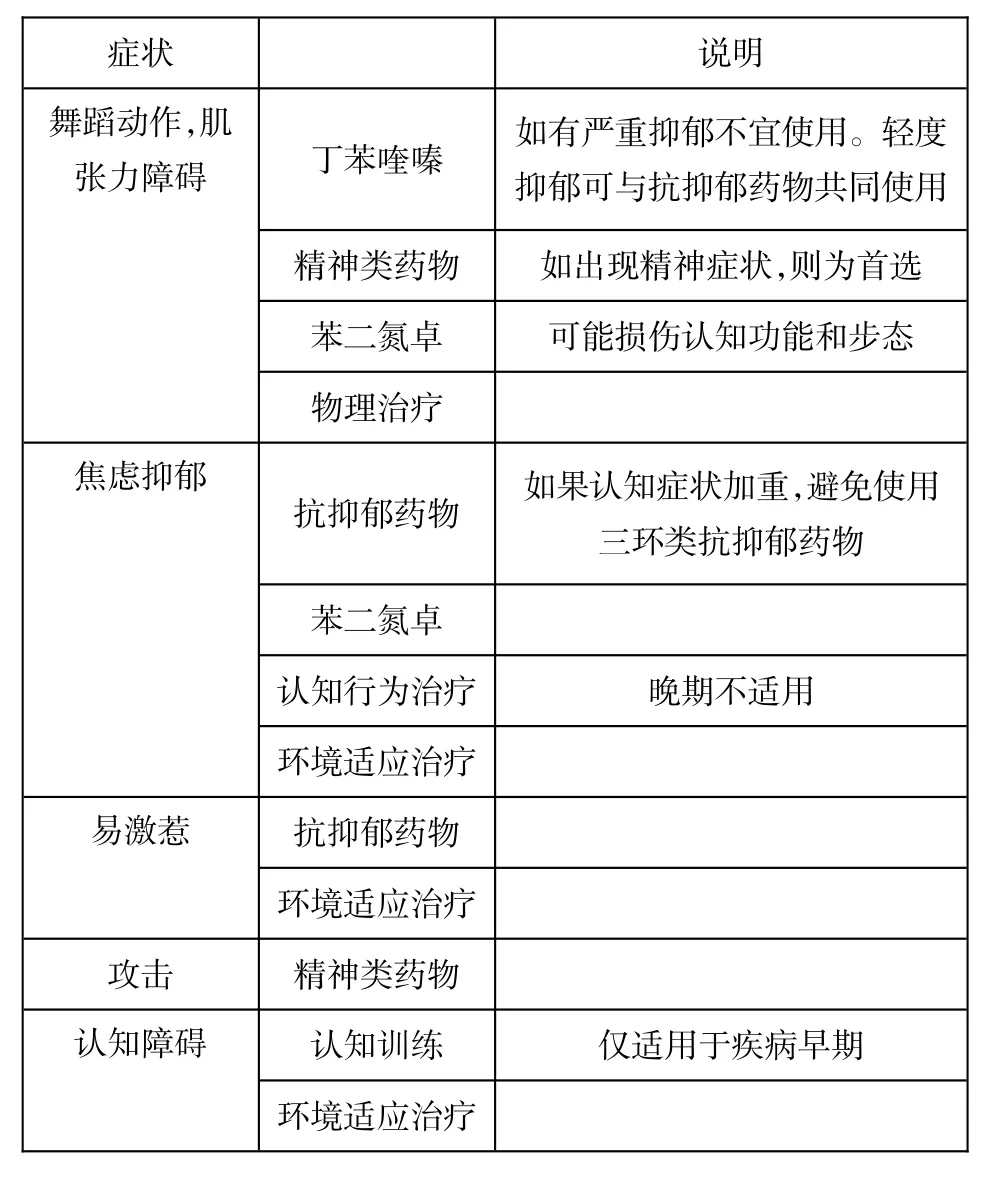
表4 HD的主要治疗形式
由于对药物治疗HD认知障碍的研究很少,目前仍未有任何药物显示有效。因此症状评估尤为重要,这样能更好地为患者和家属提供咨询,包括对自身工作的适应调整及相应对策。认知治疗可有助于患者及其周边制定相应活动计划和管理可利用资源。而环境策略(environmental strategies)对于重症病例可能有一定价值。
我们对HD特有精神症状的治疗方法仍然有限,大多数指南依赖专家意见以及其他精神症状表型为主的疾病研究。专家意见调查显示,对于强迫[45],和易激惹[46]症状,可以建议采取阶梯性治疗方法。在轻度认知障碍的患者中,可先采用五羟色胺再摄取抑制剂(serotonin reuptake inhibitor),并可同时结合行为治疗。如果同时出现抑郁,焦虑和强迫症行为,五羟色胺再摄取抑制剂也可为一线用药。对上述两组患者来说,也建议采用行为治疗,行为治疗也可减轻需要长期收容治疗的晚期患者的压力。用药调整到最适剂量后,下一步就是合并用药治疗。相似的方法也可能用于抑郁症的治疗。
吞咽困难一般出现在疾病的后期,是由于面部非自主运动障碍,运动控制能力下降,快速度进食倾向以及药物副作用,包括由抗胆碱能引起的口干而造成的。目前暂无研究可以指导治疗吞咽困难,但是已有指南对吞咽食物(应在认知障碍影响到学习能力前开始指导),正确的准备食物以及在监控环境下提供食物给出指引。医师需要提早与患者讨论胃管辅助进食,以便了解患者的选择,以及潜在并发症如窒息、呼吸道肺炎的风险。体重减轻在HD病症中很常见,这是由于吞咽困难、舞蹈症动作及新陈代谢失调所造成,所以用适当方法增加患者能量摄入非常重要。
HD患者及其亲人同时还会面对不少社会及医疗问题,这都需要受过训练的专业人士合理解决。
9 结论
HD虽是一种可怕的疾病,但如果认为治疗无望就大错特错。目前已经有不少对症疗法可用,综合看护计划也使患者及其家人受益。临床研究及看护项目也已非常到位,在世界范围内建立起了包括诊所、研究人员和医疗组织的大型网络。欧洲亨廷顿病协作网(EHDN)就是很好的代表,并已在这方面取得极大成就。EHDN最主要的患者注册研究有超过10,000 HD患者和正常对照。由CHDI赞助的全球性研究Enrol-HD,已于2012年正式启动。而在中国,医师及HD患者也共同建立了中国亨廷顿病协作网,并启动了多个合作研究项目。
同英文版)
(中山大学附属第一医院 冯璐扬翻译裴中校对)
R742.2
2015-09-25)
A
(责任编辑:李立)
10.3969/j.issn.1002-0152.2015.10.001
*瑞士伯尔尼大学神经科(University of Bern,Switzerland)
猜你喜欢
文苑(2020年8期)2020-09-09
孩子(2019年10期)2019-11-22
天津医科大学学报(2019年6期)2019-08-13
科学24小时(2019年4期)2019-06-10
医药前沿(2019年29期)2019-01-05
海峡姐妹(2018年3期)2018-05-09
中成药(2017年8期)2017-11-22
科学生活(2016年7期)2016-07-25
医学研究杂志(2015年7期)2015-06-22
郑州大学学报(医学版)(2015年1期)2015-02-27
