
Noonan综合征临床特征分析:附2例病例报告
2015-12-24 03:28王伟王春庆殷华田秦杰
生殖医学杂志 2015年5期
王伟,王春庆,殷华,田秦杰*
(1.中国医学科学院,北京协和医学院,北京协和医院妇产科,北京 100730;2.胜利油田中心医院,东营 257034;3.山东省烟台市芝罘区妇幼保健院,烟台 264000)
Noonan综合征(NS)1968年由Noonan正式提出,是一种以特殊面容、身材矮小、智力发育障碍伴先天性心脏病、骨骼发育异常、出血倾向、淋巴管发育不良为特征的多发性先天畸形,其在新生儿中发病率约为1/2 000~1/1 500[1]。大部分患者往往因为青春期第二性征不发育、月经不来潮等现象到妇产科门诊就诊,患者多表现为特殊面容、身材矮小、青春期原发性闭经、第二性征不发育等。因NS临床较为罕见,临床医生对该病认识不足,容易延误诊断及治疗。本文通过对2例NS患者的临床资料进行分析,探讨NS的诊断及治疗,以期引起临床医生的重视,减少其误诊和漏诊,达到较好的治疗效果。
临床资料
一、研究对象
2例患者均来自北京协和医院妇产科门诊,均以身材矮小、原发性闭经为主诉就诊。
二、临床特征及一般检查
病例1 患者初诊时间2001年,年龄19岁,身高138cm,学习成绩差,中途辍学。一般检查见面部多痣、内眦赘皮、眼距宽、腭弓高,盾胸、乳房不发育(按照Tanner分级评估标准[2]为TannerⅠ级),无腋毛、肘外翻、脊柱向左侧弯曲,外阴幼稚、无阴毛,尿道口与阴道口可见,肛查可扪及小子宫。
病例2 患者初诊时间2005年,年龄16岁,身高131cm,13岁开始乳房自动发育,学习成绩中等。一般检查见面部多痣、腭弓高,乳房发育(TannerⅢ级),无腋毛,肘外翻,外阴幼稚、无阴毛、尿道口与阴道口可见,肛查可扪及小子宫。
2例患者父母均非近亲结婚,父母外貌、身高、体格无异常,智力表现无异常,无遗传性家族病史,母亲孕期均无服药史。
三、辅助检查情况
对2例患者行染色体核型、性激素水平、妇科B超、骨龄及骨密度、甲状腺功能等辅助检查发现,2例患者外周血淋巴细胞染色体核型均为46,XX,促甲状腺激素(TSH)轻度升高;B 超检查可探及小子宫、小卵巢(表1),B 超显示:病例1 卵巢内可见大小不等的卵泡,病例2卵巢内未见明显卵泡。病例1 骨龄检查显示11 岁,明显滞后于实际年龄(19岁),且骨密度明显下降;病例2骨龄检查显示16岁与实际年龄(16岁)基本相符,骨密度正常。卵泡刺激素(FSH)、黄体生成素(LH)、雌二醇(E2)等性激素水平检测显示,病例1表现为低促性腺激素性闭经,病例2各激素水平基本正常(表1)。
四、诊断
患者均为女性,有类似先天性卵巢发育不全又称特纳(Turner)综合征的临床表现,如青春期第二性征不发育、原发性闭经、身材矮小、面部多痣、腭弓高、肘外翻等。本研究中例1还可见内眦赘皮、骨骼异常、智力稍低下等。临床上对存在特殊体征、青春期女性第二性征不发育或发育欠佳,无阴毛、腋毛,内外生殖器发育幼稚;有子宫及阴道,采用雌孕激素序贯人工周期治疗可来月经;性激素水平检测提示FSH、LH、E2显著低于正常水平或基本正常;染色体核型检查为46,XX,即可诊断为NS。NS患者骨龄表现可落后于实际年龄或与实际年龄基本相符,骨密度低于同龄正常水平或基本正常。
五、治疗
病例1因骨龄明显落后于实际年龄,骨骺未愈合,故起初给予具有雌激素、孕激素和雄激素3种激素活性的替勃龙(利维爱片,欧加农,荷兰)2.5mg/d口服治疗2年半,身高增长4.5cm,复查骨龄片示骨骺闭合。后予雌孕激素序贯治疗即每周期口服克龄蒙(拜耳,德国)21d,1 片/d,或每周期口服芬吗通(苏威,荷兰)28d,1片/d,促进第二性征发育,建立规律月经,并予口服福善美片(阿仑磷酸钠片,默沙东,美国)治疗骨质疏松,70mg/次,1次/周,服用3个月后复查骨密度。病例2曾予生长激素(GH,长春金赛药业)0.066 mg·kg-1·d-1皮下注射治疗3个月,身高增长1cm,之后使用克龄蒙、芬吗通行雌孕激素序贯治疗(具体用法同病例1)。
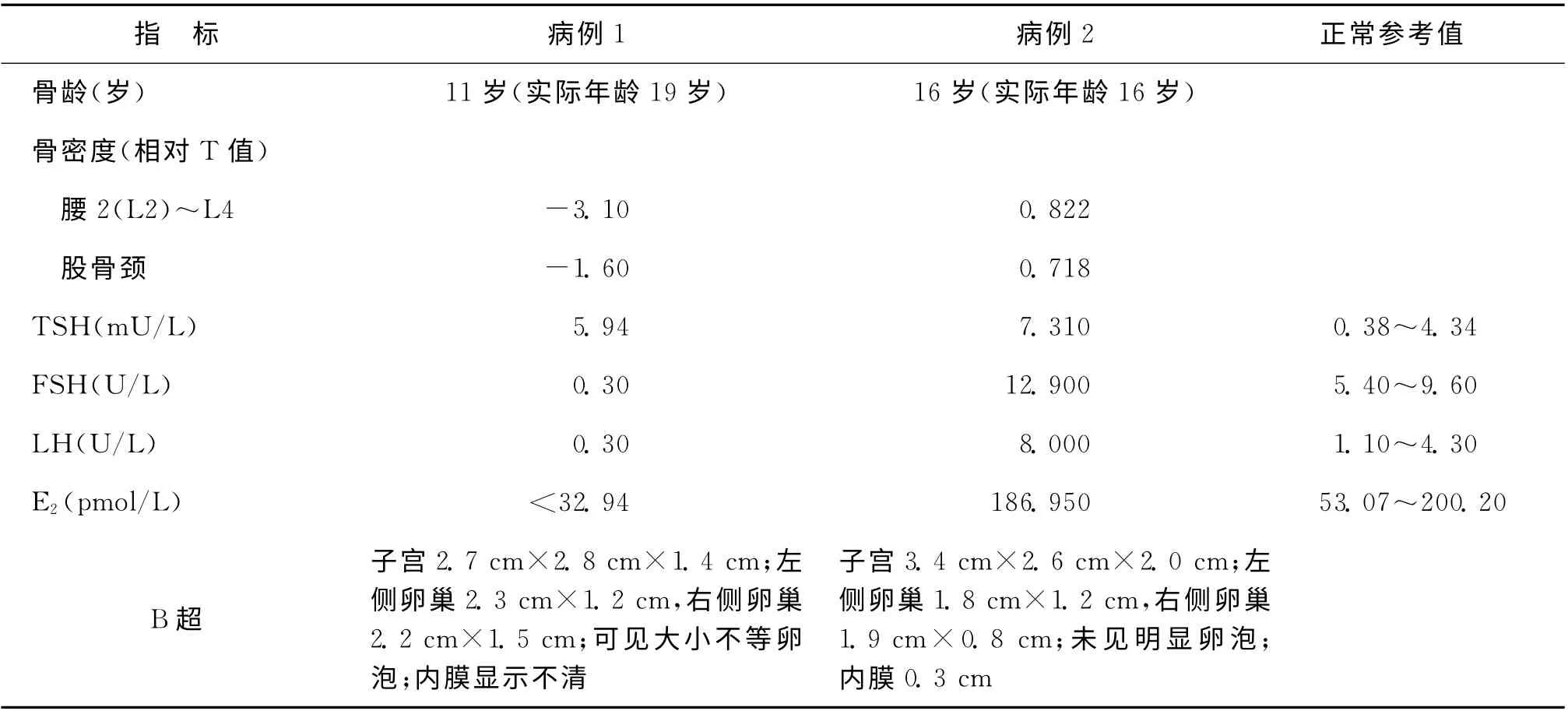
表1 两例NS患者辅助检查结果
六、随访
2例患者行雌孕激素序贯治疗后均有规律的月经来潮,病例1随访至目前31岁,最终身高为148cm;病例2随访至目前24 岁,最终身高132.8cm。第二性征均有不同程度发育,乳房发育达TannerⅣ~Ⅴ级,阴毛、腋毛无明显生长。2 例患者均已婚,未生育。
讨 论
一、NS的病因和发病机制
NS是一种临床表现多样的遗传综合征,又称先天性侏儒痴呆综合征或翼状颈综合征,家族性病例占所报道病例的30%左右,表现为常染色体显性遗传,其余大多数病例属于散发类型[3-4]。其发病机制与大鼠肉瘤蛋白/丝裂原活化的蛋白激酶(RAS/MAPK)信号通路的相关基因突变,导致该通路异常激活有关。细胞因子、生长因子等配体与细胞表面相应受体结合后,使生长因子受体结合蛋白2(GRB2)募集,与鸟苷酸交换因子(SOS)和蛋白酪氨酸磷酸酶非受体型11(PTPN11)形成复合体,活化RAS蛋白,通过一系列磷酸化反应引起快速生长的纤维肉瘤蛋白-分裂原活化抑制剂-蛋白激酶(RAF-MEK-ERK)级联效应。然后,ERK 进入细胞核内调节基因转录,从而对刺激做出适宜的短期或长期反应,在细胞生长、增殖、分化、存活及凋亡中扮演关键的角色[5]。近50%的NS为12号染色体(12q24.1)上PTPN11 基因发生错义突变,导致非受体蛋白酪氨酸磷酸酶自体磷酸化而获得自身功能所致[6]。除PTPNl1基因外,还有其他致病基因亦可导致NS发生,包括柯尔斯顿鼠肉瘤病毒癌基因同 系 物(KRAS)(<5%)[7]、交 换 因 子 同 系 物1(SOSl)(10%~13%)[8]、RAF1(3%~17%)[9-10]及原癌基因丝氨酸苏氨酸激酶(BRAF)[11]基因,显示该病较高的遗传异质性。
二、NS的临床表现与诊断
NS的临床特点为男女性均可罹患,主要见于男性,可累及多系统,临床表现有类似Turner综合征的特征,包括特殊面容,如面部多痣、上睑下垂、眼距宽、耳廓低位、腭弓高,颈短,蹼颈,后发际低等;骨骼异常:盾胸、肘外翻、脊柱侧弯等;身材矮小。Tartaglia等[3]研究报道NS患者多合并生殖器发育不良,女性可表现为卵巢发育不良,男性可有隐睾;智力轻至中度低下,或基本正常;大多数合并先天性心脏病,以右心系统为重,多见肺动脉瓣狭窄或发育不良以及肥厚性心肌病。1/3的患者可因凝血因子或血小板缺乏有出血倾向。特殊面容在出生时可不明显,儿童期表现显著,成年后可能又不再明显。此外,10%NS患者存在低频波段听力受损,25%的患者存在高频波段听力障碍。95%NS 患者可有斜视、弱势、屈光不正、白内障、眼球震颤等1种或几种表现。有家族遗传性的,由于男性病人多有隐睾而无生育能力,因此由母亲向子代传递较为多见。身材矮小是NS的主要表型之一,83%患儿身高低于第3个百分点,并影响他们的最终身高[12]。同时,有青春发育延迟表现:35%男性NS患者在13.5岁以后进入青春期,44%女性NS患者在13岁以后进入青春期[13]。青春期女性患者大多因原发闭经、第二性征不发育来妇产科就诊,一般检查可见患者乳房多不发育,阴毛、腋毛无或稀少,内外生殖器发育幼稚,有输卵管、子宫与阴道。采用人工周期后可来月经,促进乳房发育。
辅助检查:B 超可见子宫、卵巢小,有时可见卵巢内有卵泡;性激素水平测定FSH、LH 及E2显著降低或基本正常;骨密度多下降,骨龄多低于实际年龄;可合并甲状腺功能减退,TSH 升高;染色体核型检查为46,XX。
根据典型的临床表现及辅助检查,NS 诊断基本可成立。本文2例患者均符合NS诊断。病例1 FSH、LH、E2均明显降低,闭经,无乳房发育;由于缺乏生理水平的性激素刺激,骨密度显著降低,骨龄小于实际年龄。病例2则FSH 稍高,LH 正常,E2基本正常,故乳房有自行发育,骨密度基本正常,骨龄与实际年龄基本相符。原发闭经的原因可能与子宫尚未发育成熟、子宫内膜太薄(0.3cm)等有关。
三、鉴别诊断
NS与Turner综合征、LEOPARD 综合征(面部雀斑、心电图异常、眼距过宽、肺动脉瓣狭窄、外生殖器异常、生长迟缓和耳聋综合征)、心脸皮肤综合征、科斯特罗(Cestello)综合征、Aarskog综合征的症状和体征有相似部分[14],故需进行鉴别。Turner综合征临床较常见,后几种则相对罕见。虽然NS与Turner综合征有许多相似之处,如都有类似的特殊面容,骨骼异常,身材矮小;B超检查可示子宫、卵巢小;骨密度低下,骨龄低于实际年龄;常合并甲状腺功能低下等。但Turner综合征一般无家族史,多为散发病例;患者一般智力正常;绝大多数性腺发育不全,卵巢内无卵泡,FSH、LH 明显升高,达到绝经后妇女水平,E2则显著降低;心血管畸形以左心系统为主,多见主动脉瓣狭窄和主动脉缩窄[15]。而NS大多为常染色体显性遗传,有家族史;部分NS患者青春期可有正常的性发育,卵巢内可见卵泡。另外,染色体核型检查亦有助于鉴别,NS往往核型正常,为46,XX;Turner综合征则往往表现为不同类型的异常染色体核型,包括数量异常或X 染色体结构异常,如45,XO、45,X/46,XX、46,X,r(X)等,以45,XO 核型最为常见[2]。
LEOPARD 综合征以常染色体显性方式遗传。因雀斑呈年龄相关性,故在幼年期容易被误诊为NS。LEOPARD 综合征主要由PTPN11基因突变引起,其次为RAF1基因突变。
心脸皮肤综合征也有多发先天畸形、智力障碍、先天性心脏缺陷和特征性的面部表现等,但该病患者鼻尖更圆、鼻翼更宽,眉毛、睫毛稀疏,常有毛囊过度角化性丘疹等。可由BRAF、KRAS、MEK1、MEK2基因突变引起。
科斯特罗综合征,其面部特征、身材矮小、心脏异常等表现需与NS鉴别。科斯特罗综合征患者多表现为出生体重增加,鼻梁增宽,皮肤松弛,随时间推移色素沉着增加,脱发,手掌足底部皱褶深,尺骨偏斜,肛周乳头瘤,早衰。85%患者由哈维鼠肉瘤病毒癌基因同系物(HRAS)基因突变引起,对横纹肌肉瘤、神经母细胞瘤和膀胱癌易感。
Aarskog综合征,其面部特征与身材矮小的特点与NS 类似,呈X 连锁方式遗传,由FGD1(FYVE)基因突变引起,一般不合并先天性心脏病。基因测序亦有助于鉴别。
四、治疗
临床上对于NS的治疗原则一般为:一旦确诊NS即进行智力、视力、听力、生长发育及心脏等多系统的评估,并予智力训练、GH 治疗及先天性心脏病的外科治疗等,往往需要多科协作[16]。NS 的治疗目标是治疗先天性畸形,改善最终身高,促进第二性征发育,建立规律月经及减少各种并发症的发生。
对NS患者行性激素治疗的目的是促进第二性征发育,改善身高,建立规律月经,防止骨质疏松的发生,并为未来生育做准备。人体正常骨骼形成需要雌、雄激素的共同作用,因此对于确诊NS 的患者,可先行骨龄检测,对于骨骺未愈合、骨龄明显低于实际年龄的患者,可予GH、替勃龙(利维爱)等治疗。虽然NS 患者的身材矮小不是由GH 缺乏引起,但是在骨骺闭合前及时给予GH 治疗对改善身高有益。美国FDA 于2007 年批准了重组人GH用于治疗NS 患者身材矮小的治疗,推荐剂量为0.066mg·kg-1·d-1,但GH 治疗花费较高。本研究中病例1初诊时实际年龄19岁,骨龄检测相当于11岁,骨骺未闭合,但因经济困难,不能承受高额治疗花费,选择口服利维爱2.5mg/d治疗,治疗最初6个月身高增长1.5cm,共服用利维爱治疗30个月,直到复查骨龄提示相当于16~17岁,身高共增长4.5cm,最终身高148cm;病例2初诊时16岁,曾用GH 治疗3个月,身高增长1cm,但由于患者开始治疗时骨骺已基本闭合,且患者自行停药,故最终身高(132.8cm)并不理想。故建议临床上对身高矮小的患者,要结合临床特征和染色体检查结果尽早明确诊断,对确诊NS的患者要尽早积极治疗,以期有效改善最终身高。
NS合并心血管畸形的严重程度直接决定患者预后,如无严重的心血管畸形,患者寿命可与正常人相似,因此一旦发现合并心血管畸形,应积极进行手术或介入治疗,以改善患者的生活质量,延长寿命。NS伴发的其他系统畸形中,约2/3 的脊柱畸形需手术矫正[17];10%NS患者伴有肾脏畸形,但一般不需要治疗[18];因为存在出血风险,NS患者在进行有创操作前应请血液科医师会诊以使出血风险降至最低[19]。
雌激素用于NS的治疗,一方面可以刺激乳房和生殖器官的发育,同时也会促进骨骺愈合。因此临床上为了兼顾最大限度地改善患者身高和促进生殖器官发育,治疗上一般先改善身高,骨骺闭合后再开始使用雌激素。且单独使用雌激素可能导致子宫内膜增生症,增加子宫内膜癌的发病风险,而加用孕激素可消除该风险,因此临床上一般采用雌孕激素序贯周期疗法建立月经周期,该方案一般长期使用,维持治疗20~30年。
五、预后和随访
关于NS 的长期随访研究资料较少,Shaw等[20]研究发现NS患者的病死率为9%,死亡年龄从几个月至61岁不等。25%的合并肥厚性心肌病的NS患者在第1年内死于心衰,但与家族性肥厚性心肌病患者相比猝死率较低。一项关于297名荷兰NS患者的研究表明,伴PTPN11基因突变的NS患者的患癌风险是正常人群的3.5倍[21]。由于NS患者往往合并多系统器官疾病,约1/3的患者需要应用治疗心律失常或心衰的药物,甚至放置起搏器,并需长期应用雌孕激素药物等,唐卉等[22]还曾报道NS伴双眼脉络膜缺损1例,因此临床上应进行定期随访,如肝功能和肾功能,心电图、心脏彩超,听力、视力等,随访中一旦发现问题,应积极处理。本研究中两例患者均给予激素治疗,月经规律,第二性征不同程度地发育,已婚未育,定期随访未发现明显异常。
[1] Nora JJ,Nora AH,Sinha AK,et al.The Ullrich-Noonan syndrome(Turner phenotype)[J].Am J Dis Child,1974,127:48-55.
[2] 于传鑫,李儒芝 主编.妇科内分泌疾病治疗学[M].上海:复旦大学出版社,2012:249-322.
[3] Tartaglia M,Kalidas K,Shaw A,et al.PTPN11mutations in Noonan syndrome:molecular spectrum,genotype-phenotype correlation,and phenotypic heterogeneity[J].Am J Hum Genet,2002,70:1555-1563.
[4] Sharland M,Morgan M,Smith G,et al.Genetic counselling in Noonan syndrome[J].Am J Med Genet,1993,45:437-440.[5] Aoki Y,Niihori T,Kawame H,et al.Germline mutations in HRAS proto-oncogene cause Costello syndrome[J].Nat Genet,2005,37:1038-1040.
[6] Tartaglia M,Mehler EL,Goldberg R,et al.Mutations in PTPN11,encoding the protein tyrosine phosphatase SHP-2,cause Noonan syndrome[J].Nat Genet,2001,29:465-468.
[7] Schubbert S,Zenker M,Rowe SL,et al.Germline KRAS mutations cause Noonan syndrome[J].Nat Genet,2006,38:331-336.
[8] Tartaglia M,Pennacchio LA,Zhao C,et al.Gain-of-function SOS1mutations cause a distinctive form of Noonan syndrome[J].Nat Genet,2007,39:75-79.
[9] Pandit B,Sarkozy A,Pennacchio LA,et al.Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy[J].Nat Genet,2007,39:1007-1012.
[10] Razzaque MA,Nishizawa T,Komoike Y,et al.Germline gainof-function mutations in RAF1cause Noonan syndrome[J].Nat Genet,2007,39:1013-1017.
[11] Sarkozy A,Carta C,Moretti S,et al.Germline BRAF mutations in Noonan,LEOPARD,and cardiofaciocutaneous syndromes:molecular diversity and associated phenotypic spectrum[J].Hum Mutat,2009,30:695-702.
[12] Ranke MB,Heidemann P,Knupfer C,et al.Noonan syndrome:growth and clinical manifestations in 144cases[J].Eur J Pediatr,1988,148:220-227.
[13] Romano AA,Dana K,Bakker B,et al.Growth response,nearadult height,and patterns of growth and puberty in patients with noonan syndrome treated with growth hormone[J].J Clin Endocrinol Metab,2009,94:2338-2344.
[14] Roberts AE,Allanson JE,Tartaglia M,et al.Noonan syndrome[J].Lancet,2013,381:333-342.
[15] Van der Hauwaert LG,Fryns JP,Dumoulin M,et al.Cardiovascular malformations in Turner’s and Noonan’s syndrome[J].Br Heart J,1978,40:500-509.
[16] Romano AA,Allanson JE,Dahlgren J,et al.Noonan syndrome:clinical features,diagnosis,and management guidelines[J].Pediatrics,2010,126:746-759.
[17] Lee CK,Chang BS,Hong YM,et al.Spinal deformities in Noonan syndrome:a clinical review of sixty cases[J].J Bone Joint Surg Am,2001,83:1495-1502.
[18] Sharland M,Burch M,McKenna WM,et al.A clinical study of Noonan syndrome[J].Arch Dis Child,1992,67:178-183.
[19] Tofil NM,Winkler MK,Watts RG,et al.The use of recombinant factor VIIa in a patient with Noonan syndrome and life-threatening bleeding[J].Pediatr Crit Care Med,2005,6:352-354.
[20] Shaw AC,Kalidas K,Crosby AH,et al.The natural history of Noonan syndrome:a long-term follow-up study[J].Arch Dis Child,2007,92:128-132.
[21] Jongmans MC,van der Burgt I,Hoogerbruggep M,et al.Cancer risk in patients with Noonan syndrome carrying a PTPN11mutation[J].Eur J Hum Genet,2011,19:870-874.
[22] 唐卉,杨彩珍,张月玲,等.Noonan综合征伴双眼脉络膜缺损一例[J].中华医学遗传学杂志,2006,23:137.
猜你喜欢
今日农业(2022年1期)2022-06-01
保健与生活(2022年10期)2022-05-06
科学24小时(2020年5期)2020-08-02
健康之友·下半月(2020年6期)2020-07-04
小雪花·初中高分作文(2019年8期)2019-10-07
新医学(2019年4期)2019-09-10
世界中医药(2019年7期)2019-09-10
阅读(科学探秘)(2019年5期)2019-07-19
心血管外科杂志(电子版)(2018年1期)2018-11-08
中国当代医药(2017年2期)2017-03-18
