
基于高通量测序分析海带表面细菌群落结构
2017-03-14 02:52孙丕海钱坤李晓丽杨志平丁鉴锋孙雪莹马悦欣
大连海洋大学学报 2017年1期
孙丕海,钱坤,李晓丽,杨志平,丁鉴锋,孙雪莹,马悦欣
(1.大连海洋大学海珍品苗种培育基地,辽宁大连116023;2.大连金普新区海洋与渔业局,辽宁大连116100;3.大连海洋大学农业部北方海水增养殖重点实验室,辽宁大连116023;4.大连汇新钛设备开发有限公司,辽宁大连116039)
基于高通量测序分析海带表面细菌群落结构
孙丕海1,钱坤2,李晓丽3,杨志平4,丁鉴锋3,孙雪莹3,马悦欣3
(1.大连海洋大学海珍品苗种培育基地,辽宁大连116023;2.大连金普新区海洋与渔业局,辽宁大连116100;3.大连海洋大学农业部北方海水增养殖重点实验室,辽宁大连116023;4.大连汇新钛设备开发有限公司,辽宁大连116039)
为研究大连登沙河湾和正明寺海带养殖区海带Saccharina japonica表面及其周围海域海水的细菌群落结构,分别于2015年1月、2月和4月,采用Illumina Miseq PE300高通量测序平台对样本基因组DNA 的16S rRNA基因V3~V4区PCR扩增片段进行测序分析。结果表明:从两个海区健康海带和海水中获得优化序列分别为130 158条和124 658条,海带序列可归为8个门和99个属,海水序列可归为14个门和135个属;在门水平,海带首要优势门为厚壁菌门 (>69%),其次为变形菌门、蓝细菌门、放线菌门和拟杆菌门 (>1%),海水首要优势门为变形菌门 (>46%),其次为厚壁菌门、拟杆菌门、蓝细菌门和放线菌门 (>1%);在属水平,海带主要优势属为乳球菌属 (>40%)和芽孢杆菌属 (>10%),其次为 Solibacillus、假单胞菌属和节杆菌属 (>3%),海水首要优势属为弧菌属 (21.58%)和乳球菌属 (28.29%),其次为科尔韦尔氏菌属、弓形杆菌属和亚硫酸杆菌属 (>2%)。研究表明,两个海区海带细菌群落组成相似,海带和海水细菌群落组成则明显不同。
海带;细菌群落;高通量测序
海带 Saccharina japonica(原名 Laminaria japonica)[1]隶属于褐藻门、褐子纲、海带目、海带科、海带属,具有较高的经济价值,并广泛应用于食品、医药、化工等方面,是大连沿海海域大规模栽培的主要藻类之一。研究人员曾使用传统培养方法对海带表面细菌进行了分离鉴定[2-4]。Balakirev 等[5]通过克隆文库技术分析发现,太平洋西北地区广泛分布的海带典型种 (TYP)及其常见形态变种 (LON和SHA)的共生微生物群落存在显著差异,认为宿主-共生体相互作用可导致高遗传相似性形式 (TYP和LON)表型不同,细菌共生体可作为区分遗传相似藻类形式的敏感标记。而假交替单胞菌属Pseudoalteromonas、弧菌属Vibrio和盐单胞菌属Halomonas可使海带发生病害[6-9]。Bengtsson等[10]首次使用高通量测序技术,研究了Laminaria hyperborea表面细菌多样性与次级生产力的关系。但有关海带表面细菌群落的研究仍然较少。本试验中采用高通量测序技术分析了大连登沙河湾和正明寺养殖区健康海带表面和海水细菌群落结构,旨在为了解细菌与海带间的相互作用提供参考。
1 材料与方法
1.1 材料
分别于2015年1月、2月和4月从大连登沙河湾和正明寺养殖区采集健康海带孢子体和海水。
1.2 方法
1.2.1 样品采集 剪取海带叶片生长点附近叶片约10 g,经无菌海水冲洗3次,用无菌自封袋封装。海水样品经0.22 μm孔径的滤膜抽滤,将滤膜及海带样品置于冰盒,邮寄美吉生物医药科技有限公司进行检测,送样24 h内,用1 L无菌生理盐水反复冲洗样品海带,将滤液通过0.02 mm孔径的滤膜过滤。
1.2.2 DNA提取及PCR扩增 依据说明书步骤用E.Z.N.A.®water DNA试剂盒 (Omega Bio-tek, Norcross,GA,USA)提取海带及水体 (过膜后样本)的总DNA,细菌16S rRNA基因V3~V4片段的扩增引物为338F(5′barcode-ACTCCTACGGGAGGCAGCA 3′)和806R(5′GGACTACHVGGG-TWTCTAAT 3′)[11-12]。PCR反应体系 (共20 μL): 5×FastPfu缓冲液4 μL,2.5 mmol/L dNTPs 2 μL,上、下游引物各0.8 μL(5 μmol/L),FastPfu聚合酶0.4 μL,模板DNA 10 ng,用ddH2O补足至20 μL。PCR反应程序为:95℃下预变性3 min;95℃下变性30 s,55℃下退火30 s,72℃下延伸45 s,共进行27个循环,最后在72℃下再延伸10 min。每个PCR样本设置3个平行,所得产物混合后经20 g/L琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒 (AXYGEN公司)切胶回收PCR产物。
1.2.3 Illumina MiSeq测序及序列处理 将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统 (Promega公司)进行DNA定量,之后按照每个样本的测序量要求,进行相应比例的混合,按标准流程Illumina Miseq PE300测序平台进行测序。
1.3 数据处理
使用 Trimmomatic和FLASH软件对测序数据进行优化,过滤序列尾部质量值20以下的碱基,拼接序列的重叠区错配比率低于0.2,barcode错配数为0,最大引物错配数为2的优化序列用于后续分析。将序列按照97%相似性进行OTUs(operational taxonomic units)聚类[13],在聚类过程中去除UCHIME嵌合体[14],得到 OTU的代表序列,与Silva数据库 (http://www.arb-silva.de)[15]进行比对,采用RDP classifier贝叶斯算法对OTU代表序列进行分类学分析[16](http://sourceforge.net/ projects/rdp-classifier/),置信度阈值为0.7。使用Mothur 1.30.1软件 (http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity)进行rarefaction分析和单样本α多样性指数分析,包括Coverage、Ace、Chao、Shannon和Simpson指数。使用Qiime软件计算beta多样性距离矩阵,进行层次聚类分析[17],使用非加权组平均法UPGMA(Unweighted pair group method with arithmetic mean)算法构建树状结构,比较样本间细菌群落差异。利用R语言工具制作稀释曲线图、文氏图、群落结构组分图和多样本相似性树。
2 结果与分析
2.1 两个海区海带及海水细菌群落组成
将3个时间点测序数据合并分析,从大连登沙河和正明寺两个海区海带和海水中共获得254 816条优化序列。所有序列可归为531个OTUs。海带序列可归为8个门、17个纲、37个目、63个科、99个属;海水序列可归为14个门、23个纲、44个目、78个科、135个属。
海带和海水门水平细菌群落组成如图1所示。从图1可见:海带首要优势门为厚壁菌门Finnicutes,其在大连登沙河湾养殖区和正明寺养殖区所占比例分别为 69.25% (HBS)和 83.37% (HFS);次优势门为变形菌门Proteobacteria、蓝细菌门Cyanobacteria、放线菌门Actinobacteria和拟杆菌门Bacteroidetes。海水第一优势门为变形菌门,其在两个海区所占比例分别为67.88% (HWB) 和46.39%(HWF);厚壁菌门在HWF中为第二优势门,其比例为38.39%,而在HWB中比例为9.52%;其他次优势门为拟杆菌门、蓝细菌门、放线菌门和疣微菌门Verrucomicrobia。
海带和海水纲水平细菌群落组成如图2所示。从图2可见:海带首要优势纲为芽孢杆菌纲Bacilli,其在两个海区所占比例分别为69.10%(HBS) 和83.03%(HFS);次优势纲包括蓝细菌纲、γ-变形菌纲 γ-Proteobacteria、α-变形菌纲α-Proteobacteria、放线菌纲 Actinobacteria和黄杆菌纲Flavobacteriia。海水HWB中第一优势纲为γ-变形菌纲 (40.72%),γ-变形菌纲在HWF中为第二优势纲 (23.22%);HWF中第一优势纲为芽孢杆菌纲 (38.13%),在HWB中芽孢杆菌纲所占比例为9.51%;HWB和HWF的其余次优势纲包括α-变形菌纲、黄杆菌纲、蓝细菌纲、ε-变形菌纲ε-Proteobacteria、δ-变形菌纲δ-Proteobacteria、β-变形菌纲β-Proteobacteria、放线菌纲、酸微菌纲Acidimicrobiia和噬纤维菌纲Cytophagia。
海带和海水属水平细菌群落组成如图3所示。从图3可见:HBS和HFS中主要优势属为乳球菌属Lactococcus(43.61%、53.46%)和芽孢杆菌属Bacillus(11.19%、12.58%),次优势属包括Solibacillus、假单胞菌属 Pseudomonas、节杆菌属 Arthrobacter、芽孢八叠球菌属Sporosarcina、链球菌属Streptococcus、Lysinibacillus、乳杆菌属Lactobacillus、Loktanella和肉杆菌属 Carnobacterium。HWB中首要优势属为弧菌属 Vibrio(21.58%),弧菌属在HWF中的比例为6.22%,HWF中首要优势属为乳球菌属 (28.29%),所占比例为6.69%;HWB和HWF中次优势属包括科尔韦尔氏菌属Colwellia、弓形杆菌属Arcobacter、亚硫酸杆菌属Sulfitobacter、芽孢杆菌属、Loktanella、假交替单胞菌属Pseudo-alteromonas、Owenweeksia、Ulvibacter、马赛菌属Massilia、极地杆菌属Polaribacter和Bacteriovorax。
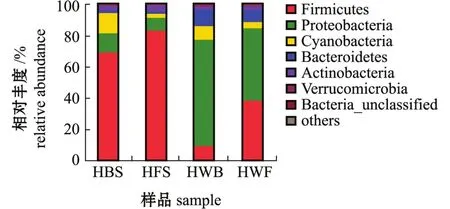
图1 两个海区海带及海水门水平细菌群落组成Fig.1 Bacterial communities in kelp and seawater in two sea areas at phylum level
2.2 海带和海水细菌群落丰度和多样性估计
海带和海水优化序列数量如表1所示,其中HBS序列66 812条,HFS序列63 346条,HWB序列62 807条,HWF序列61 851条。HBS和HFS的样本OTU分别是220和198个,而HWB和HWF的样本OTU数量高于海带,分别为408和435个。在OTU水平上生成的4个样品稀释曲线(图4)皆接近平坦,表明测序深度足够反映样本的多样性。通过Chao和Ace指数可估计海带和海水群落丰度,通过Shannon和Simpson指数可估计样本群落多样性。从表1可见,HBS和HFS群落丰度指数Chao和Ace为211~249,HWB和HWF的群落丰度指数Chao和Ace为415~451,表明海水(HWB和HWF)群落丰度高于海带 (HBS和HFS)。同样,Shannon和Simpson群落多样性指数表明,海水群落多样性也高于海带。
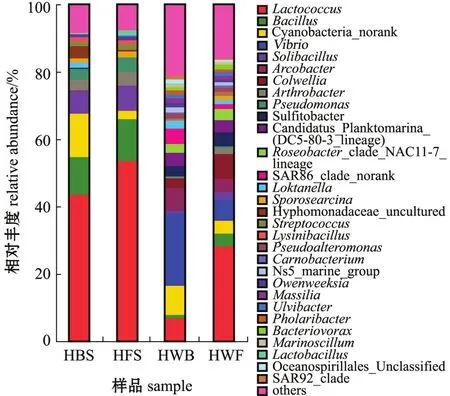
图3 两个海区海带及海水属水平细菌群落组成Fig.3 Bacterial communities in kelp and seawater in two sea areas at genus level
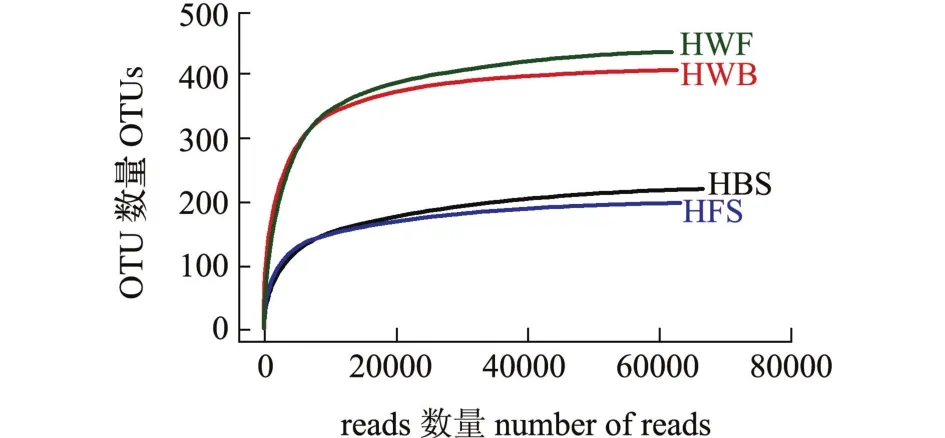
图4 两个海区海带及海水细菌群落稀释曲线Fig.4 Dilution curve of bacterial communities in kelp and seawater in two sea areas
2.3 海带和海水的细菌群落比较
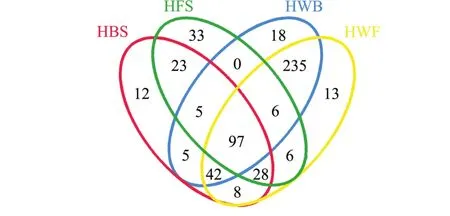
图5 两个海区海带及海水OTUs组成文氏图Fig.5 Venn diagram of OTUs shared in kelp and seawater in two sea areas
海带及海水样本所共有和独有的OTU数目见文氏图 (图5),HBS与HFS共有OTUs为153, Cyanobacteria_norank占其总 OTU的 57.74%, HWB与HWF共有OTUs为380,占其总OTU的82.07%。HBS与HWB共有OTUs为149,占其总OTU的31.11%,HFS与HWF共有OTUs为137,占其总OTU的27.62%,海带 (HBS+HFS)与海水(HWB+HWF)共有OTUs为197,占总OTU (HBS+HFS+HWB+HWF)的37.10%。聚类分析结果显示,HBS与HFS聚为一支,HWB与HWF聚为另一支 (图6)。文氏图和聚类分析结果均表明,两个海区海带之间细菌群落相似,海水之间细菌群落也相似;同一海区/两个海区的海带与海水之间细菌群落则存在明显差异。
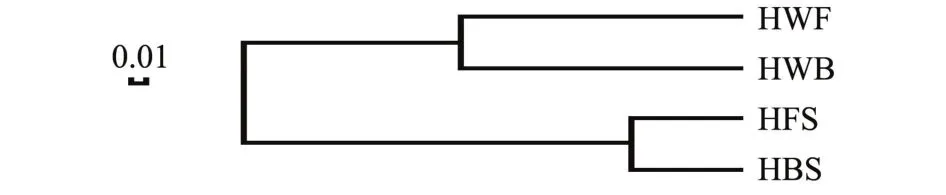
图6 两个海区海带及海水OTU水平相似树Fig.6 Similarity dendrogram of the OTU level betweenkelp and seawater in two sea areas
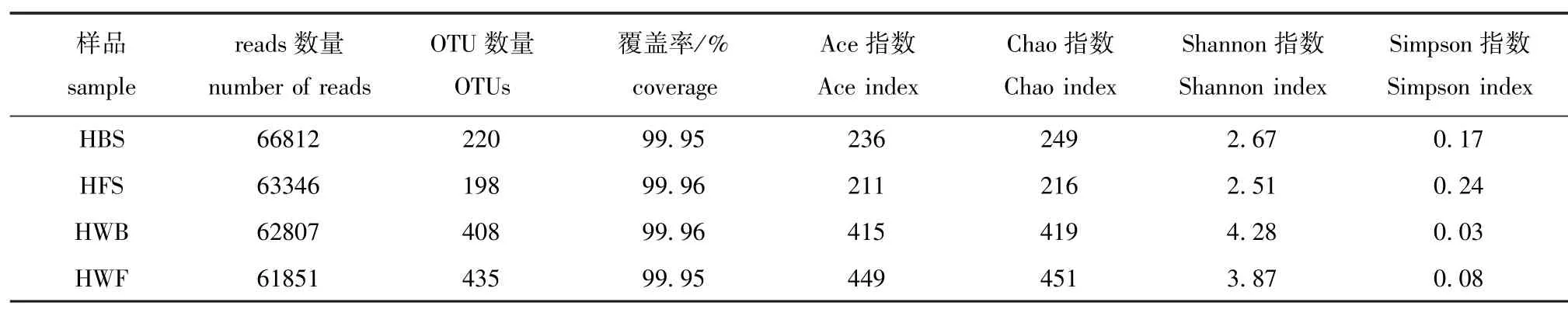
表1 两个海区海带及海水细菌群落丰富度及多样性指数Tab.1 Richness and diversity indices of bacterial communities in kelp and seawater in two sea areas
3 讨论
3.1 海带细菌群落组成分析
高通量测序结果显示,大连海区海带细菌群落包括8个门,即厚壁菌门、变形菌门、蓝细菌门、放线菌门、拟杆菌门、疣微菌门、浮霉菌门Planctomycetes和Armatimonadetes,研究人员通过培养方法和克隆文库发现,海带细菌群落由变形菌门、拟杆菌门 (CFB群)、放线菌门和厚壁菌门组成[2-5,18]。从海带属的其他种如 Laminaria hyperborea细菌群落可检测出除Armatimonadetes之外的7个门 (通过培养方法、变性梯度凝胶电泳、克隆文库、荧光原位杂交和高通量测序等方法检测)[9,19-21],从细布海带Laminaria religiosa检测的细菌群落为变形菌门 (通过培养方法检测)[22],从Laminaria longicruris细菌群落检出变形菌门和拟杆菌门 (通过培养方法检测)[23],Laminaria digitata附生细菌群落由变形菌门、放线菌门和拟杆菌门组成 (通过全细胞光谱方法检测)[24],Laminaria saccharina细菌群落也可归为变形菌门、拟杆菌门、放线菌门和厚壁菌门(通过培养方法和克隆文库方法检测)[25-26]。
在纲水平,从健康海带检测出17个纲,包括优势纲6个 (图2)。其中,γ-变形菌纲、β-变形菌纲、放线菌纲、芽孢杆菌纲和黄杆菌纲为已报道海带相关细菌群落成员[2-5,18,27]。从细布海带可分离出 γ-变形菌纲和 β-变形菌纲细菌[22],从L.longicruris分离出 γ-变形菌纲和黄杆菌纲细菌[23],从L.digitata检测出放线菌纲、γ-变形菌纲、α-变形菌纲和黄杆菌纲细菌[24],从L.saccharina检测出α-变形菌纲、β-变形菌纲、γ-变形菌纲、ε-变形菌纲、放线菌纲、黄杆菌纲和芽孢杆菌纲细菌[25-26],从L.hyperborea检测出α-变形菌纲、β-变形菌纲、γ-变形菌纲、δ-变形菌纲、蓝细菌纲、芽孢杆菌纲和放线菌纲细菌[9,19-21]。
在属水平,从健康海带中共发现99个属的细菌,包括优势属12个 (图5)。其中,Arenicella、芽孢杆菌属、棒状杆菌属Corynebacterium、黄杆菌属Flavobacterium、Granulosicoccus、假交替单胞菌属、假单胞菌属、嗜冷杆菌属、嗜冷单胞菌属Psychromonas、葡萄球菌属Staphylococcus和弧菌属为已报道海带相关细菌群落成员[2-5,18,27]。 从L.longicruris分离出黄杆菌属、假单胞菌属和弧菌属细菌[23]。从L.digitata检测出节杆菌属、副球菌属Paracoccus、假交替单胞菌属和假单胞菌属细菌[24],从L.saccharina中发现节杆菌属、芽孢杆菌属、黄杆菌属、假交替单胞菌属、假单胞菌属、鞘氨醇单胞菌属 Sphingomonas、寡养单胞菌属Stenotrophomonas、亚硫酸杆菌属和弧菌属细菌[26], 从L.hyperborea中分离出芽孢杆菌属、Granulosicoccus和Loktanella细菌[21]。
本研究中首次发现海带细菌群落成员:Armatimonadetes、蓝细菌门、浮霉菌门 Planctomycetes和疣微菌门;酸微菌纲、α-变形菌纲、拟杆菌纲Bacteroidia、梭菌纲Clostridia、蓝细菌纲、噬纤维菌纲、δ-变形菌纲、ε-变形菌纲、Negativicutes、Phycisphaerae、Sphingobacteriia和疣微菌纲 Verru-comicrobiae;优势属乳球菌属、Solibacillus、节杆菌属、芽孢八叠球菌属、链球菌属、Lysinibacillus、乳杆菌属、Loktanella和肉杆菌属。本研究结果扩展了人们对海带表面微生物群落的认识。
海带表面相关细菌组成有多方面的功能,有的是潜在的抗菌资源,如一种芽孢杆菌、一种假单胞菌和一种黄杆菌Flavobacterium sp.具有抗微生物活力[28];有的可促进植物生长,如 Pseudoalteromonas porphyrae MMM18菌株可提高萝卜、小麦、大豆、燕麦、豌豆和白菜种子的发芽率和芽长[18];但有些细菌对宿主是有害的,如Pseudoalteromonas bacteriolytica[7]和P.elyakovii[8]分别是海带红点病和叶斑点伤病害的病原菌。7株假交替单胞菌属和4株弧菌属细菌可能是海带孔烂病的潜在病原[9]。本研究从海带中发现芽孢杆菌属、假单胞菌属、黄杆菌属、假交替单胞菌属和弧菌属,其生态作用及其与宿主的相互作用有待于进一步研究。
3.2 海带和海水细菌群落比较
本试验中海带养殖区海水中的细菌群落主要由变形菌门、厚壁菌门、拟杆菌门、蓝细菌门和放线菌门组成 (图1)。这些门也分布在其他海藻生长海域水体中,如L.saccharina生长海域水体中的细菌属于变形菌门和拟杆菌门[25]。澳洲石莼生长海域水体中优势门为变形菌门、拟杆菌门和放线菌门,而蓝细菌门和厚壁菌门丰度较低。在属和OTU水平上海带细菌群落与其周围海水的细菌群落明显不同 (图3~图5)。这一结果与其他学者的研究结果类似[20,25,29]。L.saccharina和海水细菌群落的DGGE条带图谱明显不同,相似性仅为20%[14]。L.hyperborea表面生物膜与其周围海水细菌群落重叠较少[20]。在门水平澳洲石莼附生细菌群落与其周围海水细菌群落类似,而种水平两细菌群落则明显不同[29]。其原因可能是两种环境的物理差异引起群落组成有差异。海带上细菌的可能来源之一是其周围的海水,因此,海带表面的细菌可能是通过选择过程从海水较少的细菌种群中吸收或经表面直接接触细菌所致[20]。
[1] Lane C E,Mayes C,Druehl L D,et al.A multi-gene molecular investigation of the kelp(Laminariales,Phaeophyceae)supports substantial taxonomic re-organization[J].Journal of Phycology, 2006,42(2):493-512.
[2] Duan D,Xu L,Fei X,et al.Marine organisms attached to seaweed surfaces in Jiaozhou Bay,China[J].World Journal of Microbiology and Biotechnology,1995,11(3):351-352.
[3] Wang Zifeng,Xiao Tian,Pang Shaojun,et al.Isolation and identification of bacteria associated with the surfaces of several algal species[J].Chinese Journal of Oceanology and Limnology,2009,27 (3):487-492.
[4] 徐涤,秦松,庞国兴,等.青岛和大连海区海带(Laminaria japonica)外生细菌的16S rRNA基因序列分析[J].高技术通讯, 2004(2):81-86.
[5] Balakirev E S,Krupnova T N,Ayala F J.Symbiotic associations in the phenotypically-diverse brown alga Saccharina japonica[J]. PLoS One,2012,7(6):e39587.
[6] Yumoto I,Yamaguchi K,Yamada K,et al.Relationship between bacterial flora and occurrence of the Alteromonas sp.,the causative agent of red-spots on the culture bed of makonbu Laminaria japonica,in the coastal area of Funka Bay[J].Nippon Suisan Gakkaishi,1989,55(11):1907-1914.
[7] Sawabe T,Makino H,Tatsumi M,et al.Pseudoalteromonas bacteriolytica sp.nov.,a marine bacterium that is the causative agent of red spot disease of Laminaria japonica[J].International Journal of Systematic Bacteriology,1998,48(3):769-774.
[8] Sawabe T,Tanaka R,Iqbal M M,et al.Assignment of Alteromonas elyakovii KMM 162T and five strains isolated from spot wounded fronds of Laminaria japonica to Pseudoalteromonas elyakovii comb. nov.and the extended description of the species[J].International Journal of Systematic and Evolutionary Microbiology,2000,50(1): 265-271.
[9] Wang Gaoge,Shuai Li,Li Yun,et al.Phylogenetic analysis of epiphytic marine bacteria on Hole-Rotten diseased sporophytes of Laminaria japonica[J].Journal of Applied Phycology,2008,20 (4):403-409.
[10] Bengtsson M M,Sjøtun K,Lanzén A,et al.Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea[J].The ISME Journal,2012,6(12): 2188-2198.
[11] Stackebrandt E,Goodfellow M D.Nucleic Acid Techniques in Bacterial Systematics[M].Chichester:John Wiley and Sons Press,1991:115-175.
[12] McBain A J,Bartolo R G,Catrenich C E,et al.Microbial characterization of biofilms in domestic drains and the establishment of stable biofilm microcosms[J].Applied and Environmental Microbiology,2003,69(1):177-185.
[13] Edgar R C,Haas B J,Clemente J C,et al.UCHIME improves sensitivity and speed of chimera detection[J].Bioinformatics,2011, 27(16):2194-2200.
[14] Edgar R C.UPARSE:highly accurate OTU sequences from microbial amplicon reads[J].Nature Methods,2013,10(10):996-998.
[15] Quast C,Pruesse E,Yilmaz P,et al.The SILVA ribosomal RNA gene database project:improved data processing and web-based tools[J].Nucleic Acids Research,2013,41(D1):D590-D596.
[16] Wang Qiong,Garrity G M,Tiedje J M,et al.Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.
[17] Jiang Xiaotao,Peng Xin,Deng Guanhua,et al.Illumina Sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland[J].Microbial Ecology,2013,66 (1):96-104.
[18] Dimitrieva G Y,Crawford R L,Yüksel G Ü.The nature of plant growth-promoting effects of a pseudoalteromonad associated with the marine algae Laminaria japonica and linked to catalase excretion[J].Journal of Applied Microbiology,2006,100(5):1159-1169.
[19] Bengtsson M M,Øvreås L.Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea[J].BMC Microbiology,2010(10):261.
[20] Bengtsson M M,Sjøtun K,Øvreås L,et al.Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea[J].Aquatic Microbial Ecology,2010,60(1):71-83.
[21] Bengtsson M M,Sjøtun K,Storesund J E,et al.Utilization of kelp-derived carbon sources by kelp surface-associated bacteria [J].Aquatic Microbial Ecology,2011,62(2):191-199.
[22] Vairappan C S,Suzuki M,Motomura T,et al.Pathogenic bacteria associated with lesions and thallus bleaching symptoms in the Japanese kelp Laminaria religiosa Miyabe(Laminariales,Phaeophyceae)[J].Hydrobiologia,2001,445(1-3):183-191.
[23] Laycock R A.The detrital food chain based on seaweeds:I.Bacteria associated with the surface of Laminaria fronds[J].Marine Biology,1974,25(3):223-231.
[24] Salaün S,Kervarecd N,Potin P,et al.Whole-cell spectroscopy is a convenient tool to assist molecular identification of cultivatable marine bacteria and to investigate their adaptive metabolism[J]. Talanta,2010,80(5):1758-1770.
[25] Staufenberger T,Thiel V,Wiese J,et al.Phylogenetic analysis of bacteria associated with Laminaria saccharina[J].FEMS Microbiology Ecology,2008,64(1):65-77.
[26] Wiese J,Thiel V,Nagel K,et al.Diversity of antibiotic-active bacteria associated with the brown alga Laminaria saccharina from the Baltic Sea[J].Marine Biotechnology,2009,11(2):287-300.
[27] Hollants J,Leliaert F,De Clerck O,et al.What we can learn from sushi:a review on seaweed-bacterial associations[J].FEMS Microbiology Ecology,2013,83(1):1-16.
[28] Zheng Li,Han Xiaotian,Chen H M,et al.Marine bacteria associated with marine macroorganisms:the potential antimicrobial resources[J].Annals of Microbiology,2005,55(2):119-124.
[29] Burke C,Thomas T,Lewis M,et al.Composition,uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis[J].The ISME Journal,2011,5(4):590-600.
Characterization of bacterial community structure on surface of kelp Saccharina japonica by high-throughput sequencing
SUN Pi-hai1,QIAN Kun2,LI Xiao-li3,YANG Zhi-ping4,DING Jian-feng3,SUN Xue-ying3,MA Yue-xin3
(1.Seafood Seedling Breeding Base,Dalian Ocean University,Dalian 116023,China;2.Dalian Jinzhou District Oceanic and Fishery Administration, Dalian 116100,China;3.Key Laboratory of Mariculture and Stock Enhancement in North China's Sea,Ministry of Agriculture,Dalian Ocean University,Dalian 116023,China;4.Dalian Huixin Titanium Equipment Development Company Limited,Dalian 116039,China)
The genomic DNA was extracted from surface bacteria sampled from healthy kelp Saccharina japonica and surrounding seawater in the sea areas of Dengshahe bay and Zhengmingsi in Dalian in January,February and March of 2015,and a gene segment from the V3-V4 portion of the 16S rRNA gene was sequenced using Illumina Miseq PE300 high-throughput sequencing platform to examine the bacterial community on the surface of the kelp in two sea areas in coastal Dalian.A total of 130 158 and 124 658 optimized sequences were obtained from the kelp and seawater,respectively.Sequences from kelp were classified into 8 phyla and 99 genera,and sequences from seawater were identified as 14 phyla and 135 genera using 16S rRNA gene database.In the kelp community,phylum Firmicutes(>69%)was predominantly observed,followed by Proteobacteria,Cyanobacteria,Actinobacteria and Bacteroidetes(>1%).In seawater community,Proteobacteria(>46%)was most abundant,followed by Firmicutes,Bacteroidetes,Cyanobacteria and Actinobacteria.At genus level,Lactococcus(>40%)andBacillus(>10%)were dominant kelp members,followed by Solibacillus,Pseudomonas and Arthrobacter(>3%),while Vibrio(21.58%)and Lactococcus(28.29%)were the dominant members in seawater from Dengshahe bay and Zhengmingsi,respectively,followed by Colwellia,Arcobacter and Sulfitobacter(>2%).Dendrogram of hierarchical cluster analysis revealed that there were similar bacterial communities in the two sea areas,and different bacterial communities between the seawater and kelp surface.
Saccharina japonica;bacterial community;high-throughput sequencing
Q939.1
A
10.16535/j.cnki.dlhyxb.2017.01.002
2095-1388(2017)01-0007-06
2016-09-22
辽宁省自然科学基金资助项目 (2015020800);大连市金州新区海洋与渔业局项目 (2014115,2014118)
孙丕海 (1959—),男,高级工程师。E-mail:dlsph@aliyun.com
马悦欣 (1963—),女,博士,教授。E-mail:mayuexin@dlou.edu.cn
猜你喜欢
河南医学研究(2022年19期)2022-10-19
第一财经(2020年10期)2020-10-15
中国比较医学杂志(2020年4期)2020-05-26
第一财经(2020年3期)2020-02-29
第一财经(2019年11期)2019-12-02
第一财经(2019年7期)2019-07-25
水生生物学报(2019年4期)2019-07-20
生态学报(2019年11期)2019-07-08
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
