
替米沙坦抑制SOCS-3/SREBP-1c通路改善非酒精性脂肪性肝炎模型大鼠的胰岛素抵抗
2017-07-18 11:49刘复娜王素格沈毅慧蒋树林
中国实验动物学报 2017年3期
刘复娜,王素格,沈毅慧,蒋树林
(河北医科大学附属第二医院,河北省消化病研究所,石家庄 050000)
研究报告
替米沙坦抑制SOCS-3/SREBP-1c通路改善非酒精性脂肪性肝炎模型大鼠的胰岛素抵抗
刘复娜,王素格,沈毅慧,蒋树林*
(河北医科大学附属第二医院,河北省消化病研究所,石家庄 050000)
目的 探讨替米沙坦对高脂饮食诱导的非酒精性脂肪性肝炎大鼠肝组织SOCS-3通路的抑制作用及对胰岛素抵抗的干预作用。方法 70只雄性SD大鼠随机分为A(20只,正常对照)、B(30只,模型对照)和C组(20只,实验干预)。A组大鼠喂以普通饲料,B和C组喂以高脂饲料;12周末随机处理B组10只,行正葡萄糖高胰岛素钳夹试验,证实IR-NASH造模成功(100%)后,B、C两组继续高脂喂养,并予C组大鼠替米沙坦每日 5 mg/kg灌胃,16周末全部大鼠测血清IL-6、TG、TC、ALT、AST、空腹血糖(FBG)和血清胰岛素(FBI),并计算胰岛素抵抗指数;行正葡萄糖高胰岛素钳夹试验,肝组织HE肝脏病理学检查,半定量RT-PCR法检测肝组织SOCS-3和SREBP-1c mRNA表达水平。结果 高脂饮食12周大鼠进展为NASH。16周末,B组大鼠肝湿重显著高于A组,C则明显低于B组;B组出现血脂异常和IR,血清IL-6、肝脏SOCS-3 mRNA和SREBP-1c mRNA表达水平明显上调 (与A组比较P<0.01),三者分别与稳态葡萄糖输注率(VGIR60-120)显著负相关(r=0.9248,P<0.0001;r=0.9011,P<0.0001;r=0.9739,P<0.0001)。替米沙坦干预后,肝功能和血脂异常明显改善,NASH病理明显好转,SOCS-3 mRNA和SREBP-1c mRNA较B组显著下调(P<0.01),同时VGIR60-120明显增高(r=0.9532,P<0.0001;r=0.9687,P<0.0001),表明IR明显改善;而此时IL-6仍高水平,与VGIR60-120、SOCS-3、SREBP-1c失去相关关系(r=0.0071,P=0.7238;r=0.0019,P=0.8560;r=0.0002,P=0.9586),而SOCS-3 mRNA和SREBP-1c mRNA仍显著相关(r=0.9439,P<0.0001)。结论 替米沙坦干预NASH大鼠可显著减改善肝功能和IR,其机制并非抑制炎症因子IL-6,而是下调肝脏SOCS-3、SREBP-1c的表达,从而改善糖脂代谢和NASH。
非酒精性脂肪性肝炎;胰岛素抵抗;SOCS-3;SREBP-1c;替米沙坦
非酒精性脂肪性肝病/非酒精性脂肪性肝炎(nonalcoholic fat liver disease/nonalcoholic steatohepatitis, NAFLD/NASH)目前尚无疗效理想的治疗药物。抑制细胞因子信号转导抑制因子3(suppressor of cytokine signaling-3, SOCS-3)在肝脏的表达成为NAFLD药物治疗的研究热点[1-4]。尽管噻唑烷二酮(thiazolidinediones, TZD)类药物可以下调SOCS-3的表达,并显著改善胰岛素抵抗(insulin resistance, IR)[5,6],但此类药物可以引起肥胖、水肿甚至心功能不全等副作用。最近研究表明,某些ARBs类药物如替米沙坦对NAFLD/NASH具有潜在应用价值[7,8]。
鉴于IR和细胞因子等介导了NAFLD/NASH肝脏炎症及纤维化的病理过程,因此,改善胰岛素敏感性、调整细胞因子信号转导可能影响NASH的病理进程[9]。研究表明,IL-6在IR患者体内较正常组高2~3倍,可能是IR的致病介质[10];SOCS-3的过表达可引起IR和SREBP-1c的增加[3,4],并为IL-6关键性生物调节因子[11];因此推测,NASH发病机制中IL-6、SOCS-3和SREBP-1c可能存在瀑布放大关系。替米沙坦在这瀑布关系起何种作用,目前尚无研究报道。因此,本实验采用替米沙坦干预高脂饮食诱导的IR-NASH大鼠,观察对IL-6、SOCS-3和SREBP-1c的表达及其相互关系。
1 材料与方法
1.1 实验设计及NASH模型的制备
1.1.1 实验动物
选用清洁级成年雄性Sprague Dawley大鼠70只,体重(175±10) g, 4周龄,由河北医科大学实验动物学部提供【SCXK(冀)2013-1-003】。全部动物均在河北医科大学第二医院实验室【SYXK(冀)2016-003】饲养,自由进食水。准确称重、标记,适应性喂养1周。
1.1.2 分组设计及NASH模型制备
实验动物分3组:A组(正常对照)20只,给予基础饲料喂养;B组(模型对照)30只,C组(实验干预)20只,均给予配方[12]为88%基础饲料+10%猪油+2%胆固醇的高脂饲料喂养。普通饲料及高脂饲料均由河北医科大学实验动物中心配制。12周后随机选取B组10只进行正葡萄糖高胰岛素钳夹试验(euglycemic hyperinsulinemic clamp technique,EHCT),然后处死大鼠取同一部位肝组织行病理学检查。B、C组大鼠继续高脂喂养,同时C组大鼠经灌胃法给予替米沙坦每日5 mg/kg, 4周。最后全部大鼠处死,取血清及肝组织存于-70℃备用。
1.2 研究方法
自第13周始,C组给予替米沙坦每日5 mg/kg溶于1 mL 0.9%的氯化钠灌胃,A组与B组均给予等量氯化钠灌胃。给药过程于每日上午8:30~10:00完成。每周称重1次,据体重变化调节给药剂量,连续进行至16周末给药期结束,行EHCT(如前述)并进行以下研究。
1.2.1 EHCT
参照Diggs-Andrews方法[13],试验前禁食12 h,自由饮水。方法简述如下:腹腔注射2%戊巴比妥钠(60 mg/kg)进行麻醉。分离左颈静脉和右侧股动脉。左颈静脉插入留置针套管(50 U/mL肝素生理盐水溶液抗凝),结扎固定,三通管连接两台微量注射仪;右侧股动脉往近心端方向插入置套管(50 U/mL肝素生理盐水溶液)用以监测血糖。大鼠静置30 min后, 股动脉抽取血液1 mL,用罗氏血糖仪测定血糖作为基础血糖(BBG),余血置于干管备用。自颈静脉持续输注胰岛素,输注速率为每分钟10 mU/kg。每5 min后从股动脉取血测定血糖值,若低于BBG-0.5 mmol/L,则输注10%葡萄糖,输注速率从每分钟4~6 mg/kg开始,调整葡萄糖输注速率,使血糖恢复到BBG±0.5 mmol/L范围;当连续3次血糖值均在上述范围时,视为达到稳态,一般需要60 min。继续每隔5 min测血糖,必要时微调整葡萄糖输注速率,共进行至120 min。计算达稳态后60 min内平均葡萄糖输注速率VGIR60-120。
1.2.2 生化指标
16周末全部大鼠禁食水12 h后尾静脉采血,测血清甘油三酯(TG)、总胆固醇(TC)、丙氨酸氨基转移酶(ALT)、天门冬氨酸氨基转移酶(AST)和空腹血糖(FBG);采用液相平衡竞争放射免疫分析法检测血清胰岛素(FINS)和IL-6水平,以稳态模型评估法计算胰岛素抵抗指数(HOMA-IR=(FBG × FINS)/ 22.5)。
1.2.3 肝脏指数及组织学检查
将肝脏完整分离至体外,冷盐水冲洗后称量并记录肝湿重,计算肝脏指数(肝脏指数=肝湿重/体重 × 100%)。取肝右叶约1 cm × 1 cm × 0.5 cm组织固定、包埋、切片、HE染色。光学显微镜下对肝脂肪变性和炎细胞浸润程度分级[14]。肝脂肪变性占所获取肝组织标本量的范围分4度(F0-4):F0 <5%肝细胞脂肪变;F1 5%~ 30%肝细胞脂肪变;F2 31%~ 50%肝细胞脂肪变性;F3 51%~ 75%的肝细胞有脂肪变;F4 75%以上的肝细胞有脂肪变。NASH的炎症分级(G0-3):G0无炎症;G1腺泡3带呈现少数气球样肝细胞,腺泡内散在个别点灶状坏死;G2腺泡3带明显气球样肝细胞,腺泡内点灶状坏死增多,门管区轻或中度炎症;G3腺泡3带广泛的气球样肝细胞,腺泡内点灶状坏死明显,门管区轻-中度炎症伴/或门管区周围炎症。
1.2.4 SOCS-3 mRNA、SREBP-1c mRNA检测
半定量RT-PCR测肝组织SOCS-3、SREBP-1c mRNA基因转录水平的变化。主要步骤:提取肝组织总RNA,置于紫外分光光度仪上测定A260/A280在1.8~2.0之间。取RNA样品2 μL,加入6倍上样缓冲液1 μL,60 V电压电泳至溴酚蓝迁移到凝胶的3/4处;紫外灯下可见28S的宽度及亮度约是18S的两倍,且边缘清晰。引物序列检索自NCBI-Nucleotide基因库,采用Primer5.0软件设计,由北京赛百胜基因有限公司合成。SOCS-3 mRNA引物:5′-CAG CTCCAAGAGCGAGTACCAG-3′ 5′-CATGTAGTGGTGCAC CAACTTGA -3′,94℃预变性5 min,94℃变性45 s, 59℃退火45 s,72℃延伸1 min,循环35次,72℃延伸7 min,扩增产物306 bp。SREBP-1c mRNA引物:5′-GCAACACTG GCAGAGATCTACGT -3′ 5′-TGGCGGGCACTACTCAGGAA -3′,94℃预变性5 min,94℃变性30 s, 60℃退火45 s,72℃延伸1 min,循环30次,72℃延伸7 min扩增产物104 bp。GAPDH引物:5′-CCCTGAAGTACCCCATTGAA-3′ 5′-TCTCCAGGG AGGAAGAGGAT-3′,94℃预变性5 min,94℃变性45 s, 59℃退火45 s,72℃延伸45 s,循环35次,72℃延伸7 min,扩增产物516 bp。取5 μL RT-PCR产物行2%琼脂糖凝胶电泳,凝胶图像分析系统进行吸光度扫描,观察条带的灰度强弱,结果以SOCS-3或SREBP-1c和GAPDH的积分吸光度比值表示。
1.3 统计分析
2 结果
2.1 肝湿重、肝脏指数
第16周末B、C组大鼠肝湿重均显著高于A组(P<0.01);C组肝湿重显著低于B组(P<0.01)。B组与C组大鼠肝脏指数显著高于A组(P<0.01);C组肝脏指数低于B组(P<0.05)。(表1)
2.2 ALT、AST、TG、TC
第16周末,C组ALT低于B组(P<0.01);AST则与B组相比差异无显著性(P>0.05)。C组大鼠血清TC低于B组(P<0.01);而TG差异无显著性(P>0.05)。(表1)
2.3 空腹血糖、血清胰岛素、胰岛素抵抗指数
第16周末,B组大鼠FBG显著高于A组(P<0.01),C组显著低于B组(P<0.01),C组与A组相比,差异无显著性(P>0.05)。B组大鼠FINS和HOMA-IR显著高于A组与C组(P<0.01),而A组和C组差异无显著性(P>0.05)。(表1)
2.4 血清IL-6及肝组织SOCS-3 mRNA、SREBP-1c mRNA
第16周末,C组大鼠血清IL-6水平与B组差异无显著性(P>0.05),均处于较高水平,两组均高于A组(图1) (P<0.01)(图1)。A组大鼠肝组织仅有少量SOCS-3表达,B组肝组织SOCS-3表达较A组明显增强(P<0.01),C组SOCS-3表达显著下调,与A组差异无显著性(P>0.05)(图1,图2)。与SOCS-3相似, A组大鼠肝组织仅有少量SREBP-1c表达,B组肝组织SREBP-1c表达较A组显著增强(P<0.01),C组较B组显著下调(P<0.01),与A组相当(P>0.05)(图1,图2)。
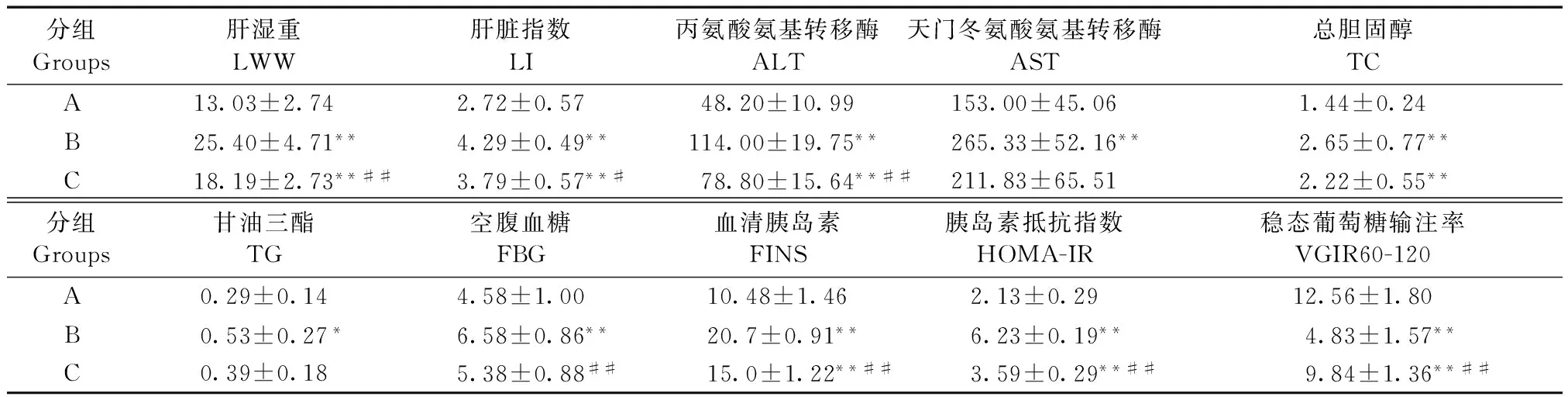
表1 16周末三组间各参数比较Tab.1 Comparison of parameters between the three groups at the end of 16th week
注:*与A比较,#与B比较:**P<0.01,*P<0.05;##P<0.01,#P<0.05。
Note.*Compared with the group A,#Compared with the group B,**P<0.01,*P<0.05;##P<0.01,#P<0.05.

图1 各组大鼠血清IL-6水平及肝组织SOCS-3、SREBP-1c mRNA的表达Fig.1 Changes of serum IL-6, expression levels of SOCS-3 mRNA and SREBP-1c mRNA in the rat livers
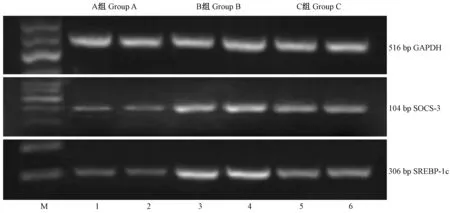
图2 肝组织SOCS-3、SREBP-1c mRNA的表达Fig.2 The expression levels of SOCS-3 mRNA and SREBP-1c mRNA in the rat livers
2.5 正葡萄糖高胰岛素钳夹试验
第16周末,B组大鼠VGIR60-120明显低于A组(P<0.01),C组则明显高于B组(P<0.01)(表1)。相关分析表明,A和B组各20只大鼠血清IL-6水平、肝组织SOCS-3 mRNA和SREBP-1c mRNA表达与葡萄糖稳态输注率呈明显相关关系(图3,图4)。而C组20只大鼠,血清IL-6仍居高不降,与VGIR60-120、SOCS-3 mRNA、SREBP-1c mRNA不呈正或负相关关系;而SOCS-3 mRNA和SREBP-1c mRNA表达,与葡萄糖稳态输注率仍分别呈明显负相关关系,SOCS-3 mRNA和SREBP-1c mRNA两者间仍呈明显正相关关系(图5)。
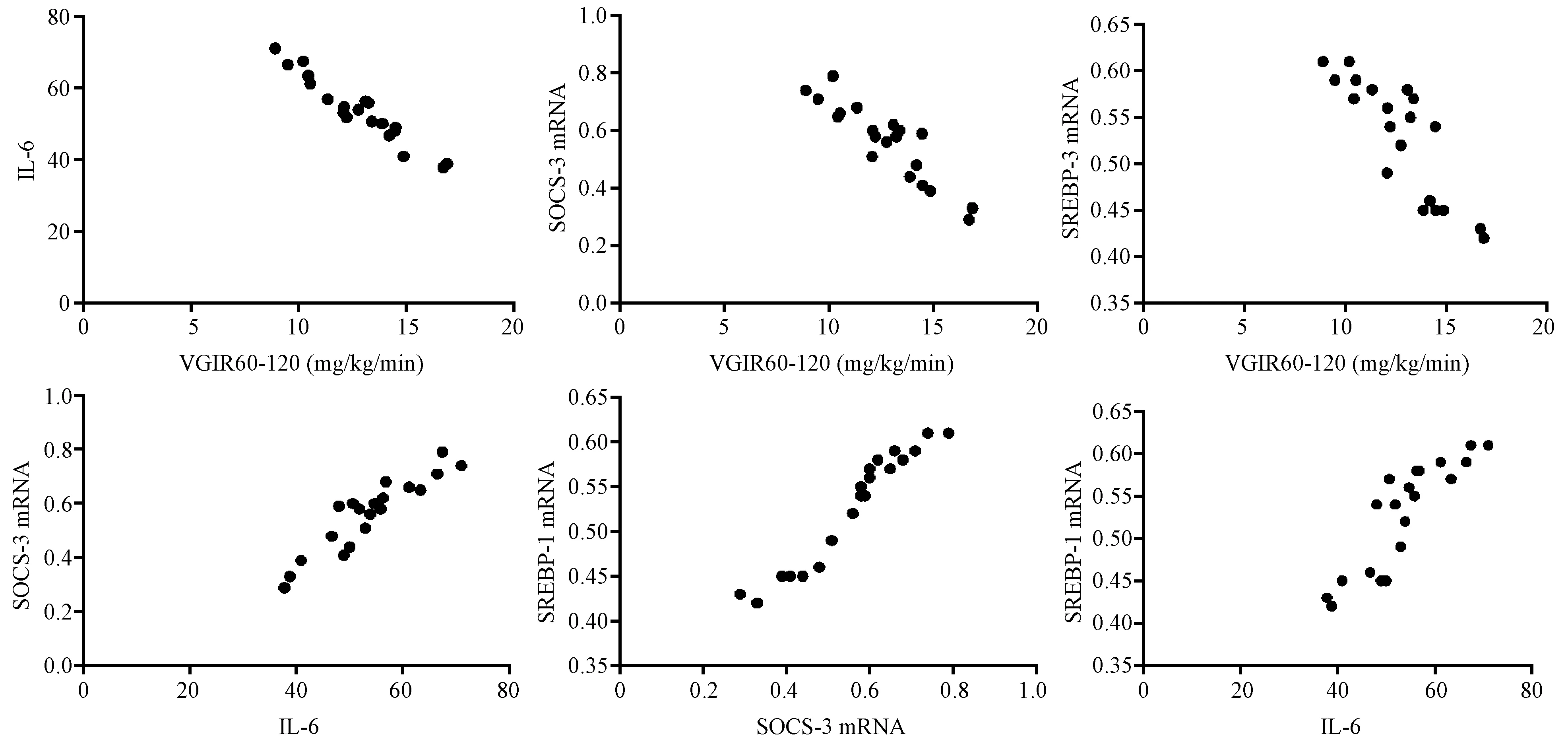
图3 A组各项指标的相关性分析Fig.3 Correlation analysis of the indexes in the group A
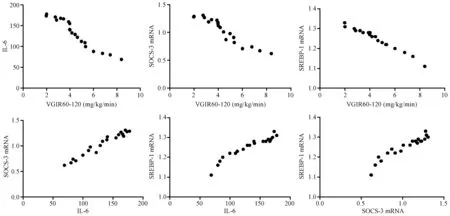
图4 B组各项指标的相关性分析Fig.4 Correlation analysis of the indexes in the group B
2.6 肝病理学改变
光镜下观察A组大鼠,肝小叶结构清晰,肝细胞中央有大而圆的核,胞质均匀,无脂滴浸润,汇管区及小叶内无炎细胞浸润。B组大鼠12周均出现弥漫性肝细胞脂肪变性和炎细胞浸润,随造模时间延长脂肪变程度逐渐加重,16周脂肪变程度均为F4;炎症分级以G2-G3为主,与正常组比较差异有显著性。C组也存在一定的肝细胞脂肪变性,但以F2为主,较B组明显改善。炎症分级以G1为主,但统计学分析较B组差异无显著性。B组16周3/20只大鼠出现中央静脉周围、窦周少量纤维组织增生,A组和C组未见明显肝组织纤维化(表2,图6)。
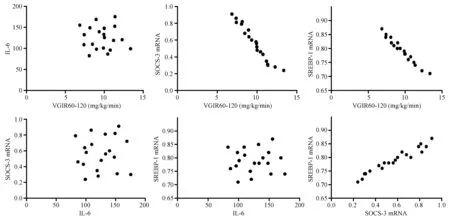
图5 C组各项指标的相关性分析Fig.5 Correlation analysis of the indexes in the group C

分组Groups肝细胞脂肪变性(F)Steatosisofhepatocytes肝组织炎症分级(G)HistologicalinflammatorygradeF0F1F2F3F4meanrankG0G1G2G3meanrankA2000005.5200005.5B00021825.35**049721.55*C25121015.65**##2106119.45*
注:*与A比较,#与B比较:**P<0.01,*P<0.05,##P<0.01。
Note.*Compared with the group A,#compared with the the group B,**P<0.01,*P<0.05,##P<0.01.
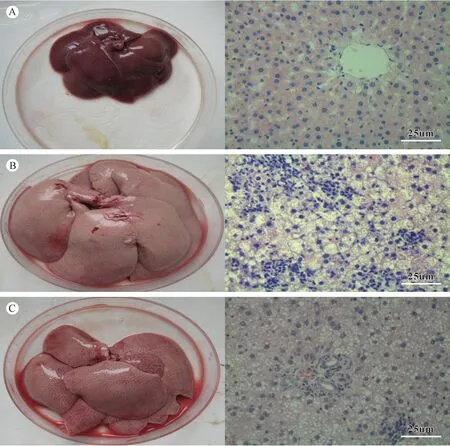
图6 全肝外观及肝组织学比较(A正常对照,B模型对照,C实验干预)Fig.6 Changes in gross appearance and histology of the rat livers(A: A nomal control rat. B: A model control rat. C: A rat of the intervention group)
3 讨论
目前普遍认为,IR在NASH的发病中起关键作用,改善IR成为治疗NASH的重要环节[15,16]。有研究发现IR患者的血清IL-6水平较正常者明显升高,与IR的发生密切相关,其分子机制可能为IL-6干扰胰岛素的信号转导,形成胰岛素受体后信号转导通路的缺陷[10,17,18];SOCS-3作为接头分子参与了IL-6介导的IR[19-21];同时上调肝脏SREBP-1c的表达[3,4],进而引起肝细胞内脂肪堆积。但IL-6、SOCS-3、SREBP-1c三者间及其与IR-NASH间的关系目前尚未阐明。
研究表明采用高脂饮食喂养SD大鼠可建立IR-NASH模型[22],本实验证实,IR-NASH大鼠血清中存在高水平的IL-6,肝组织SOCS-3亦呈高表达状态。理论上,SOCS-3的表达可能会抑制IL-6引起的IR,但本研究显示,SOCS-3的高表达不仅未能改善IR,反而使IR进一步加剧,我们称之为肝组织SOCS-3高过表达状态(overtop expression)。随着IR-NASH模型大鼠SOCS-3呈过高表达,HOMA-IR亦明显加剧;正葡萄糖高胰岛素钳夹研究表明,稳态葡萄糖输注率明显低于正常对照组,表明肝细胞胰岛素敏感性显著受损。结果提示,肝组织SOCS-3本身可能会直接通过某种途径引起IR。本研究还表明,IR-NASH模型大鼠SREBP-1c与SOCS-3均呈现高过表达状态,二者分别与稳态葡萄糖输注率呈明显相关,二者之间亦成显著相关关系。提示SOCS-3和SREBP-1c与IL-6共同参与了NASH的发病。
因此,抑制IL-6/SOCS-3/SREBP-1c可能会改善IR-NASH。NASH的治疗目前尚无理想的药物,近年来研究认为过氧化体增殖物激活受体γ激动剂—噻唑烷二酮类(TZDs) 药物对于IR-NASH的治疗有一定的帮助[20,23,24]。但TZDs类药物在降低肝脂肪沉积的同时增加了体重,造成肥胖,还可引起水钠潴留造成水肿、心衰等副作用[25],限制了该类药物在NASH患者中的应用。
研究发现,血管紧张素受体阻滞剂(ARBs)类药物替米沙坦可使高血压肥胖者在有效降压同时减轻体重[26],且替米沙坦作为AT1受体拮抗剂,降压疗效确切,又对心、脑、肾等器官具有良好的保护作用。有研究表明,替米沙坦可能具有PPARγ激动作用[27],但是否可改善糖脂代谢、改善IR尚未完全明确。替米沙坦是否对IL-6/SOCS-3/SREBP-1c介导IR途径起作用亦未见研究报道。既往关于IL-6/SOCS-3/SREBP-1c通路的研究多以脂肪细胞和平滑肌细胞为研究载体,其研究的主要目的亦限于糖尿病和心脑血管疾病,少有以肝细胞为研究载体的报道。
本研究表明替米沙坦可有效降低IR-NASH大鼠血清TG、ALT水平,显著改善NASH大鼠HOMA-IR,稳态葡萄糖输注率亦显著提高,提示机体胰岛素敏感性显著改善。肝组织学研究表明,替米沙坦干预后肝细胞脂肪变性自F4降为F2,炎症反应自G3降为G1~G2。替米沙坦作用机制为抑制肝组织SOCS-3的高过表达状态。一方面可抑制SREBP-1c的表达,抑制肝细胞对脂类的摄取减轻肝细胞脂肪变性;另一方面,改善IL-6所致胰岛素受体后缺陷,恢复或改善胰岛素敏感性。经替米沙坦干预的IR-NASH大鼠肝组织SOCS-3高过表达状态受到抑制,HOMA-IR和稳态葡萄糖输注率明显改善。
本研究还发现,替米沙坦在抑制IR-NASH大鼠肝组织SOCS-3表达时,血清IL-6水平未见明显减低。表明作为PPARγ半激动剂,替米沙坦改善NASH大鼠IR的作用靶点在SOCS-3水平,对IL-6无影响。NASH的发病机制错综复杂,可以假设适度的SOCS-3表达可负反馈抑制IL-6对胰岛素受体后通路的抑制作用,二者存在一个适度的动态平衡;而SOCS-3的高过表达则直接强化IL-6所致之胰岛素受体后缺陷,导致IR,并诱导SREBP-1c表达而加重IR和NASH进展。
总之,IL-6/SOCS-3/SREBP-1c参与了IR-NASH的发病过程;替米沙坦可明显改善IR-NASH模型大鼠的肝功能,改善IR,其机制不是抑制IL-6的分泌,而可能为下调SOCS-3和SREBP-1c表达,从而改善能量代谢和NASH。然而本研究未涉及IL-6和SREBP-1c以外与SOCSs相关的多种细胞因子(如TNF-α等)及转录因子(如chREBP),而这些因子可能都参与了NASH的发生和进展。以替米沙坦为代表的ARBs对NASH的治疗作用是否通过AT1R实现的,抑或直接发挥PPARs激动作用,本研究尚未涉及。
[1] Tilg H. The role of cytokines in non-alcoholic fatty liver disease [J]. Dig Dis, 2010,28(1): 179-185.
[2] Usui I, Tobe K. The role of inflammation in the development of insulin resistance in type 2 diabetes [J]. Nihon Rinsho, 2011,69(3): 555-562.
[3] Ueki K, Kondo T, Ronald C. Role of suppressors of cytokine signaling SOCS-1 and SOCS-3 in hepatic steatosis and the metabolic syndrome [J]. Hepatol Res,2005, 33: 185-192.
[4] Ueki K, Kondo T, Tseng YH, et al. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse [J]. Proc Natl Acad Sci U S A, 2004, 101(28): 10422-100427.
[5] Aoyama T, Ikejima K, Kon K, et al. Pioglitazone promotes survival and prevents hepatic regeneration failure after partial hepatectomy in obese and diabetic KK-A(y) mice [J]. Hepatology, 2009, 49(5): 1636-1644.
[6] Kanatani Y, Usui I, Ishizuka K, et al. Effects of pioglitazone on suppressor of cytokine signaling 3 expression: potential mechanisms for its effects on insulin sensitivity and adiponectin expression [J]. Diabetes, 2007, 56(3): 795-803.
[7] Georgescu EF. Angiotensin receptor blockers in the treatment of NASH/NAFLD: could they be a first-class option? [J]. Adv Ther, 2008, 25(11): 1141-1174.
[8] He H, Yang D, Ma L, et al. Telmisartan prevents weight gain and obesity through activation of peroxisome proliferator-activated receptor-delta-dependent pathways [J]. Hypertension, 2010, 55(4): 869-879.
[9] Schupp M, Clemenz M, Gineste R, et al. Molecular characterization of new selective peroxisome proliferator-activated receptor modulators with angiotensin receptor blocking activity [J]. Diabetes, 2005, 54: 3442-3452.
[10] Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects [J]. J Biol Chem, 2003, 278(46): 45777-45784.
[11] Serrano-Marco L, Rodríguez-Calvo R, El Kochairi I, et al. Activation of peroxisome proliferator-activated receptor-β/-δ (PPAR-β/-δ) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes [J]. Diabetes, 2011, 60(7): 1990-1999.
[12] Storlien L H,Oakes N D, Pan DA, et al.Syndrome of insulin resistance in the rat: Inducement by diet and amelioration with benfluorex [J]. Diabetes,1993, 42: 457-462
[13] Diggs-Andrews KA, Zhang X, Song Z, et al. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia [J]. Diabetes, 2010, 59: 2271-2280
[14] 徐正婕, 范建高, 王国良, 等. 高脂饮食致大鼠非酒精性脂肪性肝炎肝纤维化模型 [J]. 世界华人消化杂志, 2002, 101(47): 392-396.
[15] Bugianesi E, Moscatiello S, Ciaravella MF, et al. Insulin resistance in nonalcoholic fatty liver disease [J]. Curr Pharm Des, 2010, 16(17): 1941-1951.
[16] Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease[J]. Trends Endocrinol MeTab, 2008, 19(10): 371-379.
[17] Harford KA, Reynolds CM, McGillicuddy FC, et al. Fats, inflammation and insulin resistance: insights to the role of macrophage and T-cell accumulation in adipose tissue [J]. Proc Nutr Soc, 2011, 12: 1-10.
[18] Mitrou P, Lambadiari V, Maratou E, et al. Skeletal muscle insulin resistance in morbid obesity: the role of interleukin-6 and leptin[J]. Exp Clin Endocrinol Diabetes, 2011, 119(8): 484-489.
[19] Farrell GC. Signalling links in the liver: knitting SOCS with fat and inflammation [J]. J Hepatol, 2005, 43(1): 193-196.
[20] Lagathu C, Bastard JP, Auclair M, et al. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone [J]. Biochem Biophys Res Commun, 2003, 311(2): 372-379.
[21] Lang R, Pauleau AL, Parganas E, et al. SOCS3 regulates the plasticity of gp130 signaling [J]. Nat Immunol, 2003, 4(6): 546-550.
[22] 吴晏, 韩静, 黄黎明, 等. 高脂喂养合并小剂量链脲佐菌素建立2型糖尿病大鼠模型 [J].中国实验动物学报, 2012, 20(2): 11-15.
[23] Mori Y, Murakawa Y, Okada K, et al. Effect of troglitazone on body fat distribution in type 2 diabetic patients [J]. Diabetes Care, 1999, 22: 908-912.
[24] Giusti V, Verdumo C, Suter M, et al. Expression of peroxisome proliferator-activated receptor-gamma1 and peroxisome proliferator-activated receptor-gamma2 in visceral and subcutaneous adipose tissue of obese women [J]. Diabetes, 2003, 52: 1673-1676.
[25] Promrat K, Lutchman G, Uwaifo GI, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis [J]. Hepatology, 2004, 39(1): 188-196.
[26] Sugimoto K, Qi NR, Kazdová L, et al. Telmisartan but not valsartan increases caloric expenditure and protects against weight gain and hepatic steatosis [J]. Hypertension, 2006, 47(5): 1003-1009.
[27] Maejima Y, Okada H, Haraguchi G,et al. Telmisartan, a unique ARB, improves left ventricular remodeling of infarcted heart by activating PPAR gamma [J]. Lab Invest, 2011, 91(6): 932-944.
Telmisartan improves insulin resistance in the rats with nonalcoholic steatohepatitis by SOCS-3/SREBP-1c pathway
LIU Fu-na, WANG Su-ge, SHEN Yi-hui, JIANG Shu-lin*
(The Second Hospital, Hebei Medical University, Research Institution of Gastrointestinal Diseases,Shijiazhuang 050000,China)
Objective To evaluate the effects of telmisartan by SOCS-3/SREBP-1c pathway and its efficacy of improving insulin resistance (IR) in rats with high-fat diet-induced nonalcoholic steatohepatitis (NASH). Methods A total of 70 SD male rats were assigned randomly into 3 groups: A (normal control, 20 rats, basic diet), B (model control, 30 rats, high-fat diet) and C (treatment with telmisartan, 20 rats, high-fat diet). After the IR-NASH model was made successfully, proved by 10 rats randomly from the group B with euglycemic hyperinsulinemic clamp technique (EHCT) and liver histology, the rats in the group C were intragastrically administrated telmisartan (5 mg/kg/d) for 4 weeks, and then all rats were tested with EHCT and sacrificed to test the blood chemistry, interleukin-6, homeostasis model assessment of insulin resistance, hepatic pathological analysis, and semiquantitative RT-PCR for determining SOCS-3 and SREBP-1c mRNA. Results Rats with high-fat diet developed steatohepatitis and insulin resistance at the 12th week and had more weight gain and higher liver index at the 16th week. IL-6, SOCS-3 and SREBP-1c mRNA expressions in the group B were up-regulated obviously, and each was positively correlated with the velocities of glucose infusion rates at 60~120 min. Blood chemistry and pathological observation in the group C were all improved; both SOCS-3 and SREBP-1c mRNA were down-regulated, and each negatively correlated with VGIR60-120, while serum IL-6 stayed at a high level. Conclusions Telmisartan can remarkably improve hepatic function and insulin resistance in rats with IR-NASH, the mechanisms of which would not be by path of reducing the secretion of IL-6, but by down-regulating the expressions of SOCS-3 and SREBP-1c mRNA.
Nonalcoholic steatohepatitis; Insulin resistance; SOCS-3; SREBP-1c; Telmisartan; Rats
JIANG Shu-lin. Email: shulinjiang1@aliyun.com
刘复娜(1982-),女,硕士研究生,现工作于华北石油管理局总医院。研究方向:非酒精性脂肪性肝病。Email: liufuna123@163.com
蒋树林(1965-),男,教授,研究方向:慢性肝病、幽门螺杆菌及消化内镜。Email: shulinjiang1@aliyun.com
Q95-33
A
1005-4847(2017)03-0281-08
10.3969/j.issn.1005-4847.2017.03.009
2016-09-19
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
药学与临床研究(2022年4期)2022-09-14
世界最新医学信息文摘(2021年12期)2021-06-09
中华养生保健(2020年5期)2020-11-16
中国实用医药(2019年7期)2019-07-01
环球时报(2019-04-03)2019-04-03
中国生殖健康(2018年1期)2018-11-06
医学研究杂志(2015年12期)2015-06-10
中西医结合心血管病电子杂志(2014年1期)2014-08-11
