
全外显子组测序发现CSPP1所致Joubert综合征1例
2018-05-25 08:14罗敏娜刘志翠曹宗富孙殿荣马斯禹高华方李玉堂
中国计划生育学杂志 2018年1期
罗敏娜 刘志翠 曹宗富 孙殿荣 马斯禹,3 高华方 马 旭 李玉堂*
1. 国家卫生计生委科学技术研究所(北京,100081);2. 青岛市妇女儿童医院;3. 北京协和医学院研究生院
Joubert综合征(简称JS,MIM#213300)是一种罕见的神经系统疾病,发病率约为1:100 000[1]。Joubert综合征的临床表现主要有小脑蚓部发育不良或缺如、肌张力低、新生儿期的阵发性呼吸过度或呼吸暂停、眼球运动障碍、共济失调、发育迟缓和认知缺陷,部分患儿有视网膜发育不良、多囊肾和多指(趾)症、肝纤维囊肿等伴发症[2]。Joubert综合征通常由MRI结合临床表现进行确诊,其主要的影像学特点有:小脑蚓部部分或完全缺如,表现出“磨牙征”(MTS),“中线裂”或“蝙蝠翼”,其中“磨牙征”是Joubert综合征诊断的最重要特征[3]。已有的研究表明Joubert综合征是常染色体隐性遗传病,仅在因OFD1所致的Joubert综合征的家庭表现为X连锁隐性遗传模式。目前已报道的Joubert综合征致病基因有30个,分别为:INPP5E,TMEM216,AHI1,NPHP1,CEP290,TMEM67,RPGRIP1L,ARL13B,CC2D2A,OFD1,TTC21B,KIF7,TCTN1,TMEM237,CEP41,TMEM138,C5orf42,TCTN3,ZNF423,TMEM231,CSPP1,PDE6D,KIAA0586,TCTN2,CEP104,KIAA0556,B9D1,MKS1,TMEM107以及ARMC9[4-10]。这些基因绝大部分参与了纤毛的功能以及相关的信号途径调控,是纤毛相关疾病的典型代表[11-12]。本研究采用全外显子组测序技术对一个中国Joubert综合征家系的先证者DNA进行检测,分析测序数据并按照美国医学遗传学和基因组学学院(ACMG)的解读规则[13]进行解读,在父母中进行Sanger测序验证,发现了CSPP1基因上的2个杂合突变位点:c.1132C>T和c.2244_2245delAA,这两个复合杂合突变位点是Joubert综合征的致病原因。
1 材料与方法
1.1 实验材料
用于高通量测序分析的血液样本取自青岛市妇女儿童医院收治的Joubert综合征患者及其家属。全血基因组DNA提取试剂盒购自QIAGEN公司,Taq酶、DNA marker购自全式金公司,PCR扩增引物由华大六合科技有限公司合成。本课题研究所有的样本采集及实验流程均已通过国家卫生计生委科学技术研究所伦理审查委员会的审查,本人及家属均签署知情同意书。血样的采集及家系、临床资料的收集符合知情同意的原则。
1.2 DNA提取和全外显子组测序
按照QIAamp DNA Blood Mini Kit(Qiagen,德国)的操作说明从全血中提取基因组DNA,利用Qubit2.0(Invitrogen)进行定量。取0.5 μg待测样品DNA用Bioruptor NGS超声仪(Diagenode,比利时)随机打断成200 bp的片段,分别用Truseq Nano DNA Library Prep Kit (Illumina,美国) 和SeqCap EZ Exome +UTR Library(96M) Kit(Roche,瑞士)进行建库和全外显子组捕获,在Hiseq 2500测序仪上(Illumina,美国)进行2×125双端测序。测序过程由安诺优达公司完成。
1.3 测序数据分析
得到的数据去除污染的、低质量的数据后,以GRCh37.1/hg19基因组为参考基因组,依次使用Burrows-Wheeler Aligner(BWA)软件、Genome Analysis Toolkit(GATK)软件和ANNOVAR软件对所有的变异位点进行比对、分析和注释。将全部变异位点与dbSNP137数据库、千人基因组数据库(1000 Genomes, 1000g, http://www.1000genomes.org)、外显子组测序项目数据库ESP6500(http://evs.gs.washington.edu/EVS)以及Exome Aggregation Consortium数据库(http://exac.broadinstitute.org)进行比对分析和过滤筛选,去除位于内含子中的突变以及同义突变,去除MAF≥0.01的变异位点,优先对所有Joubert综合征已知致病基因的变异位点进行分析,筛选出无义突变、移码突变、剪切位点突变、终止密码子丢失等对蛋白质功能影响较大的变异位点,并结合已有文献报道,确定候选的致病基因变异位点。
1.4 Sanger测序验证及突变功能分析
对候选的致病基因突变位点,针对其所在外显子设计上下游引物,以患儿及其父母的基因组DNA为模板进行聚合酶链式(PCR)反应,并利用Sanger测序法进行验证。PCR引物序列及退火温度列表如下。PCR扩增反应产物经琼脂糖凝胶电泳检测后由ABI 3730完成Sanger测序;使用Chromas软件查看Sanger测序峰图,用SeqMan软件比对Sanger测序结果。
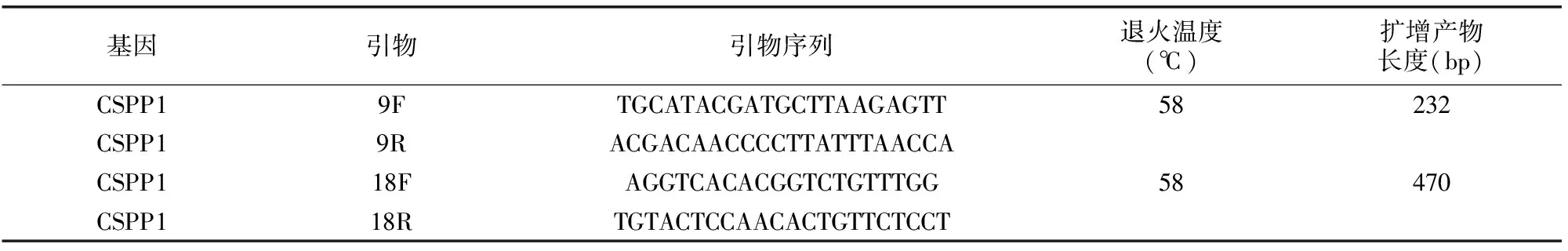
表1 CSPP1致病位点扩增引物
2 结果
2.1 临床表现
患儿女,现年4岁,因生后至今走路不稳来诊。患儿自幼发育落后,约1岁多独坐,现能独走,不稳;双手不灵活,现持勺进食撒漏多,不会正确持笔;语言认知落后,近2岁说话,至今仅会单词、少数短句表达,不认颜色且不识数。患儿系第一孕第一产,足月剖宫产,无宫内窘迫,出生时无产伤、无窒息,Apgar评分10分。家族史无特殊。查体:表情淡漠,听从简单指令,头围49cm,面容无特殊,眼球运动不协调,心肺腹无异常,四肢肌张力低,肌力大致正常,双膝反射可引出,立位双足外翻,可独走,步态不稳,蹒跚状。辅助检查:Gesell发育测查:大运动36分,精细运动49分,应物能57分,语言57分,应人能62分;染色体核型分析46,XX;血氨基酸及尿有机酸代谢筛查无明显异常;肝脏、胆囊、脾脏、双肾、输尿管及膀胱超声未见异常;脑电图正常;颅脑MRI,轴位T1WI中脑呈“磨牙征”,小脑半球于中线处靠近,可见线状裂隙,呈“中线裂征”(图1A),小脑下蚓部缺如、上蚓部发育不良,小脑上脚增粗(图1B),第四脑室上部呈蝙蝠翼状。诊断:Joubert综合征。
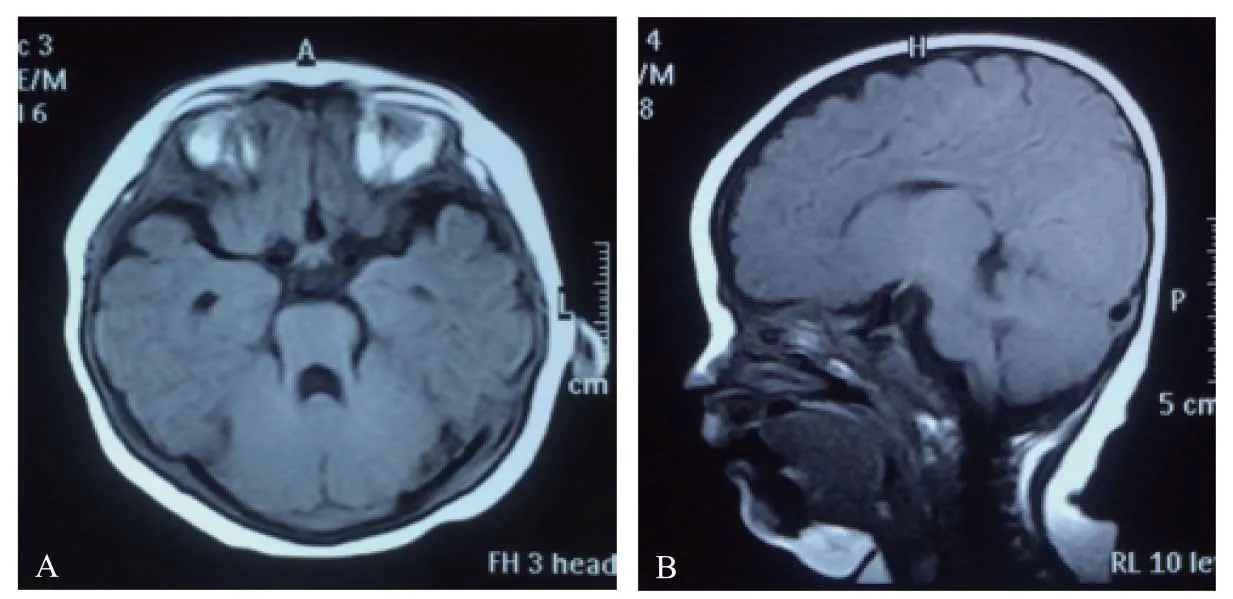
A:轴位T1WI显示“磨牙征”和“中线裂征”。B:矢状位T1WI显示小脑下蚓部缺如、上蚓部发育不良,小脑上脚增粗。
图1患儿头部MRI影像图
2.2 全外显子组测序分析
全外显子组测序产出原始数据为10.9 G,目标捕获区平均测序深度为51.26×,≥1×测序深度下的覆盖度为96.88%,≥4×测序深度下的覆盖度为95.47%,≥10×测序深度下的覆盖度为92.99%,≥20×测序深度下的覆盖度为87.46%,数据量符合实验设计要求。
在经dbSNP137、ESP6500、千人基因组数据库,ExAC数据库的过滤筛选以后,在已知的30种致病基因中发现了8号染色体上CSPP1基因(NM_024790.6)的1个点突变位点及1个InDel位点。其中,点突变位点位于9号外显子区的第1132位,由胞嘧啶突变为胸腺嘧啶(c.1132C>T),可导致其蛋白质翻译在第378位氨基酸位点发生提前终止,产生截短的蛋白质(p.R378X)。该位点在dbSNP数据库中的编号为rs374703898,在ExAC中数据库中显示其突变频率为0.00003,ClinVar数据库将其定义为致病性突变(Allele ID:214262),且已被HGMD数据库收录(CM140106)。InDel位点是在18号外显子区的2244_2245位碱基发生了缺失(c.2244_2245delAA),可导致其蛋白质翻译在第750位谷氨酸变成甘氨酸,并产生移码,于第780位终止编码(p.E750GfsX30),产生截短的蛋白质。该位点在dbSNP数据库中的编号为rs587777139,在ExAC中数据库中显示其突变频率为0.0001085,ClinVar数据库将其定义为致病性突变(Allele ID:106546),且已被HGMD数据库收录(CD140092)。
2.3 PCR扩增产物 Sanger测序验证
Sanger测序结果表明,先证者的无义突变位点c.1132C>T来自于父亲,而移码突变位点c.2244_2245delAA来自于母亲,符合常染色体隐性遗传共分离规律(图2)。上述两个位点在过去的文献中已有报道[9,14-15]。由此判定,CSPP1基因的c.1132C>T以及c.2244_2245delAA的复合杂合突变为该Joubert综合征患儿的致病性突变。
3 讨论
Joubert综合征最早于1969年被发现,是一种罕见的神经系统疾病,除了影像学MRI中的“磨牙征”表现[16]以外,其临床诊断标准有以下4项:①小脑蚓部发育不全;②肌张力低;③发育迟缓;④异常呼吸或者异常眼运动[1,17]。由于Joubert综合征的临床表现具有很强的异质性,目前国际上将所有具有“磨牙征”的疾病统称为“Joubert综合征及相关疾病”(JSRD),并根据并发症的不同将其分为6大类:①单纯型Joubert综合征;②Joubert综合征合并眼部缺陷;③Joubert综合征合并肾缺陷;④Joubert综合征合并眼肾缺陷;⑤Joubert综合征合并肝损害;⑥Joubert综合征合并口面指(趾)缺陷[4,18]。本文报道的家系先证者,小脑蚓部“磨牙征”清晰可见,小脑下蚓部缺如,上蚓部发育不良,四肢肌张力低,发育落后,符合Joubert综合征的基本特点。同时,该患儿无多指、无肝肾损伤、无眼组织缺损,考虑为单纯型的Joubert综合征。
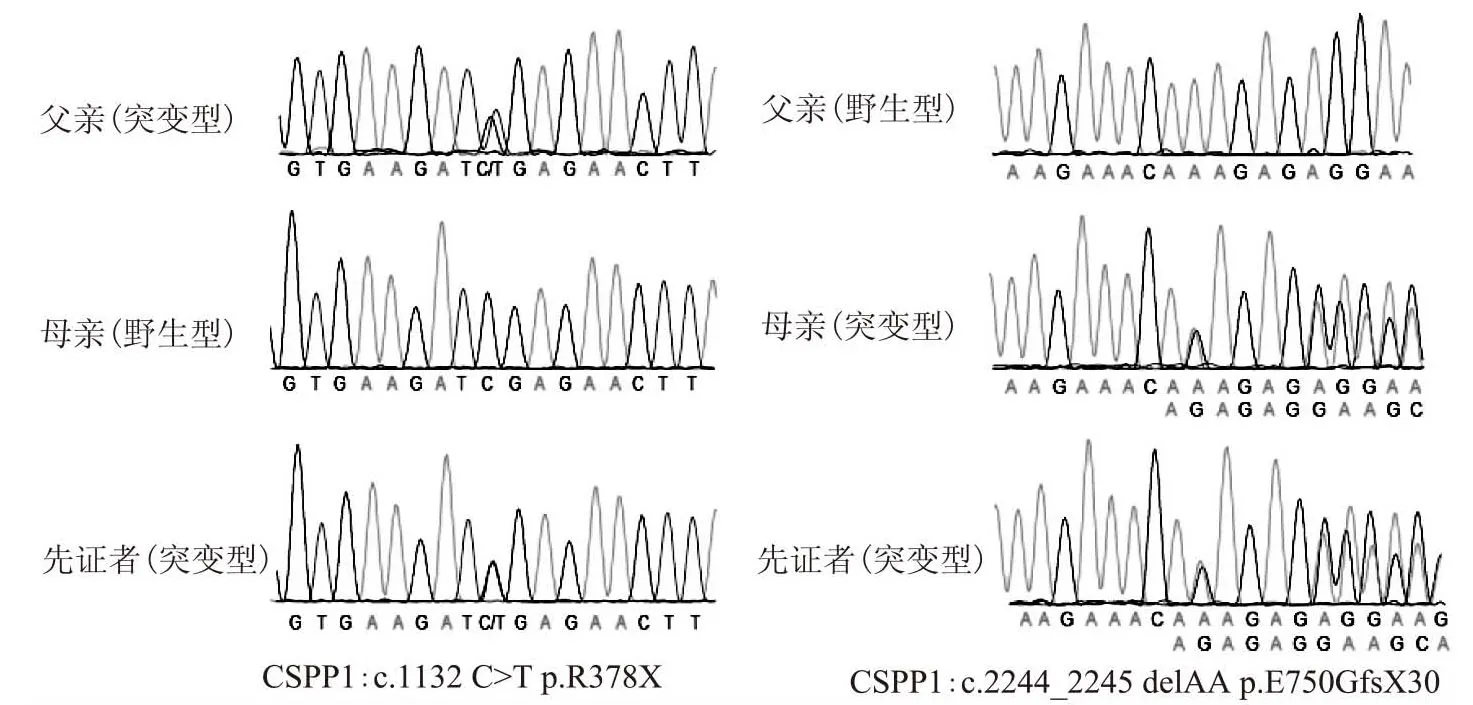
A:c.1132C>T位点的Sanger测序峰图;B:c.2244_2245delAA位点的Sanger测序峰图
图2 Sanger测序验证结果图
目前国内已报道Joubert综合征病例70余例[19-20],已明确致病基因及位点的有5例,致病基因分别是CC2D2A[21-22]、CEP290[23]、OFD[24]以及C5orf42[25],均采用高通量测序技术进行检测。全外显子测序技术是近年来在遗传性疾病检测中应用最广泛的高通量测序技术[26]。鉴于目前已经有30个Joubert综合征致病基因被发现,且仍然有约40%的病例未在已知的致病基因中发现突变,因此我们选用了全外显子组测序法进行检测。所得测序数据经过标准分析流程,按照ACMG的解读规则[13]对变异位点进行解读,最终在已知的致病基因CSPP1中筛选到了候选的致病突变位点。经PCR扩增及Sanger测序验证,并在父母中进行比对和验证,证实了CSPP1的c.1132C>T,p.R378X以及c.2244_2245delAA,p.E750GfsX30位点是该家系的致病突变位点。有研究认为CSPP1相较其它Joubert综合征基因更容易表现出截短突变致病的特点[9],本文所报道的两个位点均产生截短翻译的蛋白,与此研究相符。
CSPP1是第21个被发现的Joubert综合征致病基因。2014年,Akizu等[14]在对287名Joubert综合征患儿的外显子组测序中发现有6名先证者携带有CSPP1的截短突变并且在家系中符合遗传分离规律,因此确定CSPP1是Joubert综合征的致病基因,他同时还发现,CSPP1突变造成的Joubert综合征患儿通常仅有神经系统的表现,没有或者很少有肾脏损害、肝损害或者多指等其他伴发症,仅在2名患儿中存在听力损伤的现象,因此倾向于认为CSPP1与单纯型的Joubert综合征相关;几乎同时,在另一个研究中,Tuz等在19名纤毛疾病的患儿中检测到CSPP1的截短突变,这些患者都具有Joubert综合征典型的“磨牙征”表型,同时还有一些患者具有窒息性胸廓发育不良的表现,比如钟形胸廓,短肋骨等骨骼发育异常现象[15]。而Shaheen等[27]在另外的2个加拿大哈特人的近亲家系中检测到3名患儿具有CSPP1的截短突变,其中有2名胎儿具有与Meckel综合征相类似的表现。此后,在阿拉伯国家、韩国、日本等均发现因CSPP1突变而导致的遗传性疾病案例[10,28-29]。这些研究结果提示CSPP1突变所导致的各个疾病之间表型存在很大的差异[30]。
在模式动物中的机制研究表明,CSPP1基因的缺失会导致严重的纤毛发生缺陷。morpholino敲除cspp1的斑马鱼胚胎会表现出与其他斑马鱼Joubert综合征模型一致的表型,比如体型弯曲、肾囊肿和小脑畸形等,CSPP1是通过降低纤毛标记蛋白Arl13b的纤毛内定位来实现其作用的[15]。有研究者通过RT-PCR的方法来比较成人的大脑、皮质、小脑、胼胝体、脑桥以及胎儿的大脑、皮质、小脑以及肝脏、心脏、肾脏中CSPP1的表达量水平,发现CSPP1在神经组织中有大量表达,其中胎儿和成人的小脑中表达量最高[14]。此外,从CSPP1突变的Joubert综合征患者身上分离的成纤维细胞也表现出纤毛数量减少或长度变短等特点,并且检测到纤毛标记蛋白ARL13B在纤毛内定位的减少,这些研究结果表明CSPP1通过对纤毛功能的影响导致了疾病的发生[14-15]。
本文采用全外显子组测序技术,结合Sanger测序验证,检测到了Joubert综合征家系中CSPP1基因的两个突变(c.1132C>T和c.2244_2245delAA),是国内首次报道由CSPP1基因突变引起的Joubert综合征病例。这一研究结果为将来可能进行的产前诊断提供了分子基础。
参考文献
[1] Joubert M, Eisenring JJ, Robb JP, et al.Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation [J].Neurology,1969,19:813-825.
[2] Maria BL, Boltshauser E, Palmer SC, et al.Clinical features and revised diagnostic criteria in Joubert syndrome [J].J Child Neurol,1999,14:583-590; discussion 590-581.
[3] Maria BL, Quisling RG, Rosainz LC, et al.Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance [J].J Child Neurol,1999,14:368-376.
[4] Mancini F, Romani M, Micalizzi A, et al.Molecular Genetics of Joubert 077.2014.
[5] Bachmann-Gagescu R, Phelps IG, Dempsey JC, et al.KIAA0586 is Mutated in Joubert Syndrome [J].Hum Mutat,2015,36:831-835.
[6] Sanders AA, de Vrieze E, Alazami AM, et al.KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome [J].Genome Biol,2015,16:293.
[7] Lambacher NJ, Bruel AL, van Dam TJ, et al.TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome [J].Nat Cell Biol,2016,18:122-131.
[8] Van De Weghe JC, Rusterholz TDS, Latour B, et al.Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish [J].Am J Hum Genet,2017,101:23-36.
[9] Bachmann-Gagescu R, Dempsey JC, Phelps IG, et al.Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity [J].J Med Genet,2015,52:514-522.
[10] Vilboux T, Doherty DA, Glass IA, et al.Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center [J].Genet Med,2017,19:875-882.
[11] Reiter JF, Leroux MR.Genes and molecular pathways underpinning ciliopathies [J].Nat Rev Mol Cell Biol,2017,18:533-547.
[12] Braun DA, Hildebrandt F.Ciliopathies [J].Cold Spring Harb Perspect Biol. 2017,9.
[13] Richards S, Aziz N, Bale S, et al.Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J].Genet Med,2015,17:405-424.
[14] Akizu N, Silhavy JL, Rosti RO, et al.Mutations in CSPP1 lead to classical Joubert syndrome [J].Am J Hum Genet. 2014,94:80-86.
[15] Tuz K, Bachmann-Gagescu R, O'Day DR, et al.Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy [J].Am J Hum Genet,2014,94:62-72.
[16] Maria BL, Hoang KB, Tusa RJ, et al."Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation [J].J Child Neurol,1997,12:423-430.
[17] Saraiva JM, Baraitser M.Joubert syndrome: a review [J].Am J Med Genet,1992,43:726-731.
[18] Brancati F, Dallapiccola B, Valente EM.Joubert Syndrome and related disorders [J].Orphanet J Rare Dis,2010,5:20.
[19] 李玉堂, 陈军, 胡丽娜.Joubert综合征二例报道 [J].中国优生与遗传杂志. 2010,18:125.
[20] 李滨, 岳云龙, 金延方等.Joubert综合征的MRI及DTI表现[J].临床放射学杂志,2012,31:446-448.
[21] Chen Z, Wang JL, Tang BS, et al.Using next-generation sequencing as a genetic diagnostic tool in rare autosomal recessive neurologic Mendelian disorders [J].Neurobiol Aging,2013,34:2442 e2411-2447.
[22] 陈曦, 李胜利, 文华轩,等.Joubert综合征及相关疾病的产前及产后影像学研究 [J].中华医学超声杂志,2014,11:508-513.
[23] Wang L, Yang Y, Song J, et al.Two novel mutations in the C-terminal region of centrosomal protein 290 (CEP290) result in classic Joubert syndrome [J].J Child Neurol,2015,30:772-776.
[24] 孟晨, 张开慧, 马静,等.OFD1基因突变引起的Joubert综合征10型一例临床和基因分析 [J].中华儿科杂志,2017,26:131-134.
[25] 罗敏娜, 曹宗富, 陈军,等.全外显子组测序发现中国Joubert综合征家系C5orf42基因的新突变 [J].生殖医学杂志,2017,26:464-469.
[26] Tsurusaki Y, Kobayashi Y, Hisano M, et al.The diagnostic utility of exome sequencing in Joubert syndrome and related disorders [J].J Hum Genet,2013,58:113-115.
[27] Shaheen R, Shamseldin HE, Loucks CM, et al.Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans [J].Am J Hum Genet,2014,94:73-79.
[28] Ben-Salem S, Al-Shamsi AM, Gleeson JG, et al.Mutation spectrum of Joubert syndrome and related disorders among Arabs [J].Hum Genome Var,2014,1:14020.
[29] Suzuki T, Miyake N, Tsurusaki Y, et al.Molecular genetic analysis of 30 families with Joubert syndrome [J].Clin Genet,2016,90:526-535.
[30] Ben-Omran T, Alsulaiman R, Kamel H, et al.Intrafamilial clinical heterogeneity of CSPP1-related ciliopathy [J].Am J Med Genet A,2015,167A:2478-2480.
猜你喜欢
广西林业科学(2022年6期)2023-01-16
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
自然杂志(2022年3期)2022-08-18
医学研究生学报(2021年4期)2021-12-02
临床与实验病理学杂志(2021年3期)2021-04-25
中国生殖健康(2020年4期)2021-01-18
中华皮肤科杂志(2019年5期)2019-06-24
中国生殖健康(2018年4期)2018-11-06
